Hello Avogadro
Community, I hope you’re well:).
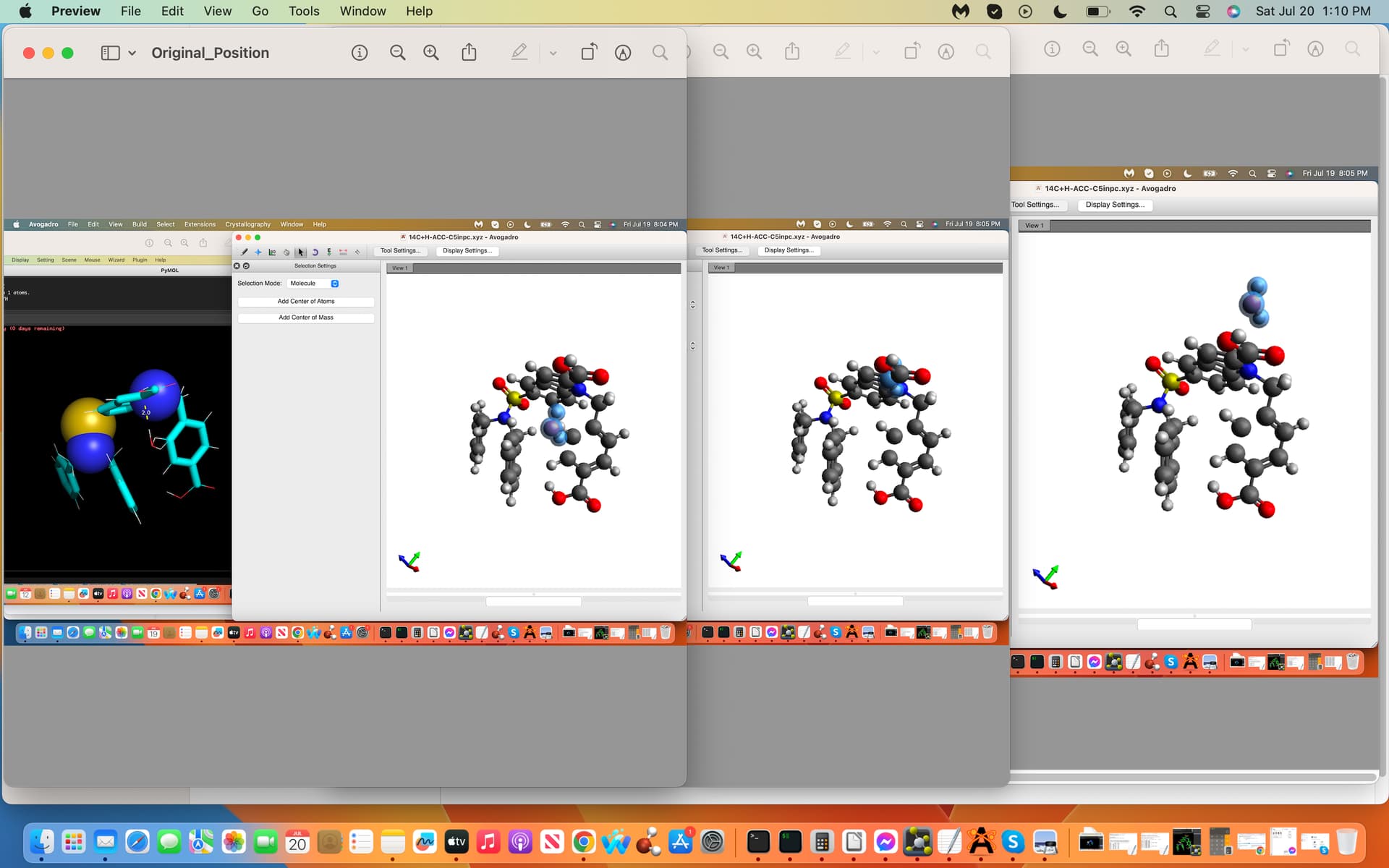
Towards the above-topic, my trajectory files .inp File´s water coordinates
need to be flipped to the other side of the ligand C5´s interaction-site to
preclude inappropriate interactions with the adjacent C-atom and to
moreover bring the final .xyz trajectory file distance to less than 5.0 A
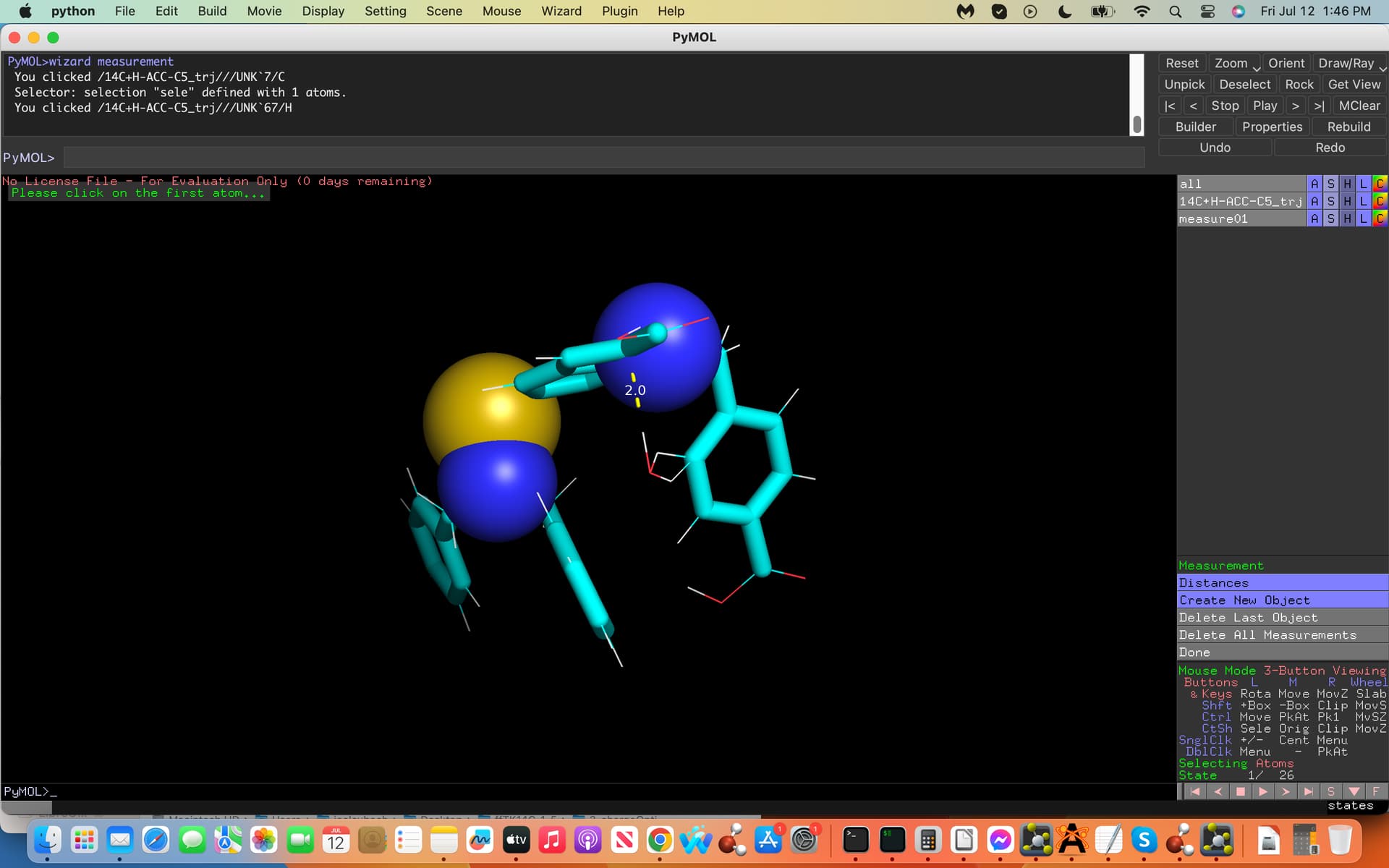
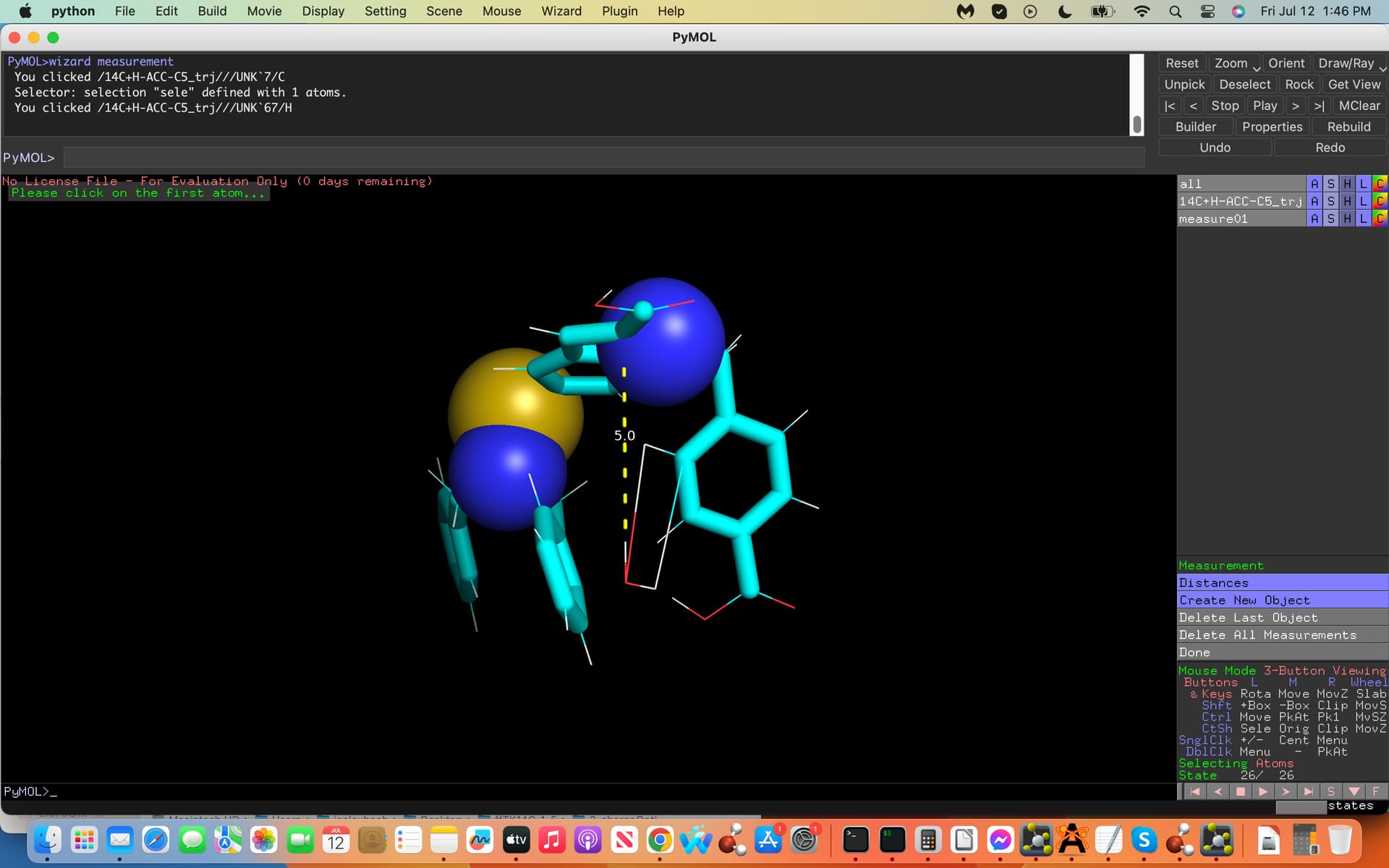
(see screenshots attached labeled .inp and .xyz, first and last coordinates
of the trajectory file, dashed lines exhibit water H-bonds to C5). How
would I flip these Cartesian Coordinates to the other side of C5?
Thanks if you know:),
Joel 
I can’t say I 100% understand what you want. But it sounds like you want to move the water molecule.
It’s fairly easy to select the water molecule atoms in Avogadro, and then use the manipulate tool to move along the xyz axes to a new position. (You don’t have to do this with the mouse, you can use the dialog to do it exactly.)
What I’d suggest is that you figure out roughly where “the other side of the ligand C5 interaction site” would be. For example, what’s the current Cartesian position of the C5? What’s the current Cartesian position of the O atom on the water?
Hello ghutchis thank you for your kind update:).
And yes I would need to place the water molecule at the same exact mirrored cartesian coordinates from the C5 atom on the opposite side (see-screenshot).
Accordingly is there a program and or technique towards properly placing this water in this exact way? Below is the cartesian coordinate file with the water at the end.
! HF 6-31G* TightSCF opt
%maxcore 1000
%geom
ConnectFragments
{ 1 2 O 6 66 }
end
Constraints
{ B 6 66 C }
{ D * 67 6 * C }
end
invertConstraints true
end
%coords
CTyp xyz
Charge 0
Mult 1
Units Angs
coords
N(1) 111.3489990234375 110.1510009765625 61.86199951171875
N(1) 106.43699645996094 106.68199920654297 61.29399871826172
C(1) 110.01799774169922 109.9229965209961 61.590999603271484
C(1) 109.39399719238281 109.42900085449219 62.775001525878906
C(1) 107.40599822998047 109.0989990234375 61.492000579833984
C(1) 108.05599975585938 108.99800109863281 62.71500015258789
C(1) 111.58399963378906 109.78700256347656 63.18000030517578
C(1) 112.3290023803711 110.26799774169922 60.78200149536133
C(1) 110.4010009765625 109.36699676513672 63.775001525878906
C(1) 109.33100128173828 110.0719985961914 60.37699890136719
C(1) 105.52200317382812 105.63999938964844 61.81399917602539
C(1) 107.18199920654297 106.31099700927734 60.084999084472656
C(1) 108.00900268554688 109.6520004272461 60.3390007019043
C(1) 112.28500366210938 109.05400085449219 59.874000549316406
C(1) 104.43399810791016 105.23699951171875 60.854000091552734
C(1) 108.29199981689453 105.3479995727539 60.439998626708984
C(1) 112.89299774169922 109.94999694824219 63.82699966430664
C(1) 111.86100006103516 107.80699920654297 60.349998474121094
C(1) 112.68299865722656 109.1719970703125 58.53499984741211
C(1) 103.25800323486328 105.99700164794922 60.75899887084961
C(1) 109.04000091552734 105.53700256347656 61.611000061035156
C(1) 104.59500122070312 104.12000274658203 60.02299880981445
C(1) 108.62300109863281 104.29000091552734 59.58100128173828
C(1) 111.8030014038086 106.7040023803711 59.49800109863281
C(1) 112.6500015258789 108.06900024414062 57.683998107910156
C(1) 112.19100189208984 106.83300018310547 58.15700149536133
C(1) 102.26399993896484 105.64399719238281 59.845001220703125
C(1) 110.08300018310547 104.66400146484375 61.93199920654297
C(1) 103.5989990234375 103.76599884033203 59.106998443603516
C(1) 109.67500305175781 103.41999816894531 59.89699935913086
C(1) 102.43399810791016 104.53099822998047 59.01499938964844
C(1) 110.40699768066406 103.6050033569336 61.07500076293945
C(1) 112.1729965209961 105.68399810791016 57.19599914550781
O(1) 105.05999755859375 108.38899993896484 62.54199981689453
O(1) 105.2770004272461 108.66000366210938 60.01300048828125
O(1) 112.87000274658203 109.46099853515625 65.09400177001953
O(1) 113.89299774169922 110.45700073242188 63.33100128173828
O(1) 111.33599853515625 104.6510009765625 57.4900016784668
O(1) 112.86000061035156 105.6510009765625 56.19200134277344
S(1) 105.84700012207031 108.26599884033203 61.30799865722656
H(1) 113.76699829101562 109.62799835205078 65.4520034790039
H(1) 110.76799774169922 104.86900329589844 58.255001068115234
H(1) 107.5479965209961 108.57099914550781 63.577999114990234
H(1) 113.30899810791016 110.40399932861328 61.242000579833984
H(1) 112.10299682617188 111.1780014038086 60.21200180053711
H(1) 110.29499816894531 109.07499694824219 64.81199645996094
H(1) 109.81300354003906 110.46700286865234 59.48699951171875
H(1) 105.10600280761719 106.01899719238281 62.750999450683594
H(1) 106.16300201416016 104.78199768066406 62.053001403808594
H(1) 107.62100219726562 107.21900177001953 59.65800094604492
H(1) 106.51899719238281 105.88300323486328 59.32099914550781
H(1) 107.4209976196289 109.73799896240234 59.42900085449219
H(1) 111.55500030517578 107.68900299072266 61.38800048828125
H(1) 113.01399993896484 110.13600158691406 58.152000427246094
H(1) 103.125 106.86199951171875 61.40599822998047
H(1) 108.78199768066406 106.35900115966797 62.27399826049805
H(1) 105.4990005493164 103.51599884033203 60.10499954223633
H(1) 108.04000091552734 104.12899780273438 58.67499923706055
H(1) 111.48600006103516 105.74800109863281 59.90700149536133
H(1) 112.96499633789062 108.14900207519531 56.64699935913086
H(1) 101.35600280761719 106.23799896240234 59.779998779296875
H(1) 110.64700317382812 104.81099700927734 62.85100173950195
H(1) 103.73200225830078 102.8949966430664 58.46900177001953
H(1) 109.91500091552734 102.59600067138672 59.229000091552734
H(1) 101.65699768066406 104.25800323486328 58.30400085449219
H(1) 111.21399688720703 102.9209976196289 61.327999114990234
H(2) 112.03199768066406 107.91600036621094 62.6349983215332
O(2) 112.24600219726562 107.0199966430664 62.374000549316406
H(2) 112.14800262451172 107.01699829101562 61.422000885009766
end
end
I still don’t think I understand what you mean by the mirrored coordinates on the opposite side. To me, it looks like you want to displace the water some distance along the black arrow, e.g. towards that carboxylic acid.
Let’s take that as an example. Say you want to place it somewhere near the carboxylic acid.
So for example, you might use the Align Tool, click the sulfur atom as the new origin, and the C of that carboxylic acid as the x-axis. Once you align, you can displace the water along the new x-axis until it’s past the carboxylic acid.
If you mean by “mirror” that there’s some sort of mirror plane, you’d probably want to define a centroid at the mirror plane (e.g., select some relevant carbon atoms, use “Add Centroid”) then subtract the Cartesian position of the oxygen atom and the centroid to figure out the vector that you sketched in black.
Then you’d select the water atoms and use the Manipulate tool to move the water along the displacement vector twice … once to move to the centroid, one to move to the new position.
If that’s not what you mean, please use like a 2D sketch or something. I’m sure it makes sense to you, but it’s unclear from your images what you mean by the C5 carbon (since we don’t know anything of your structure) or mirrored point.
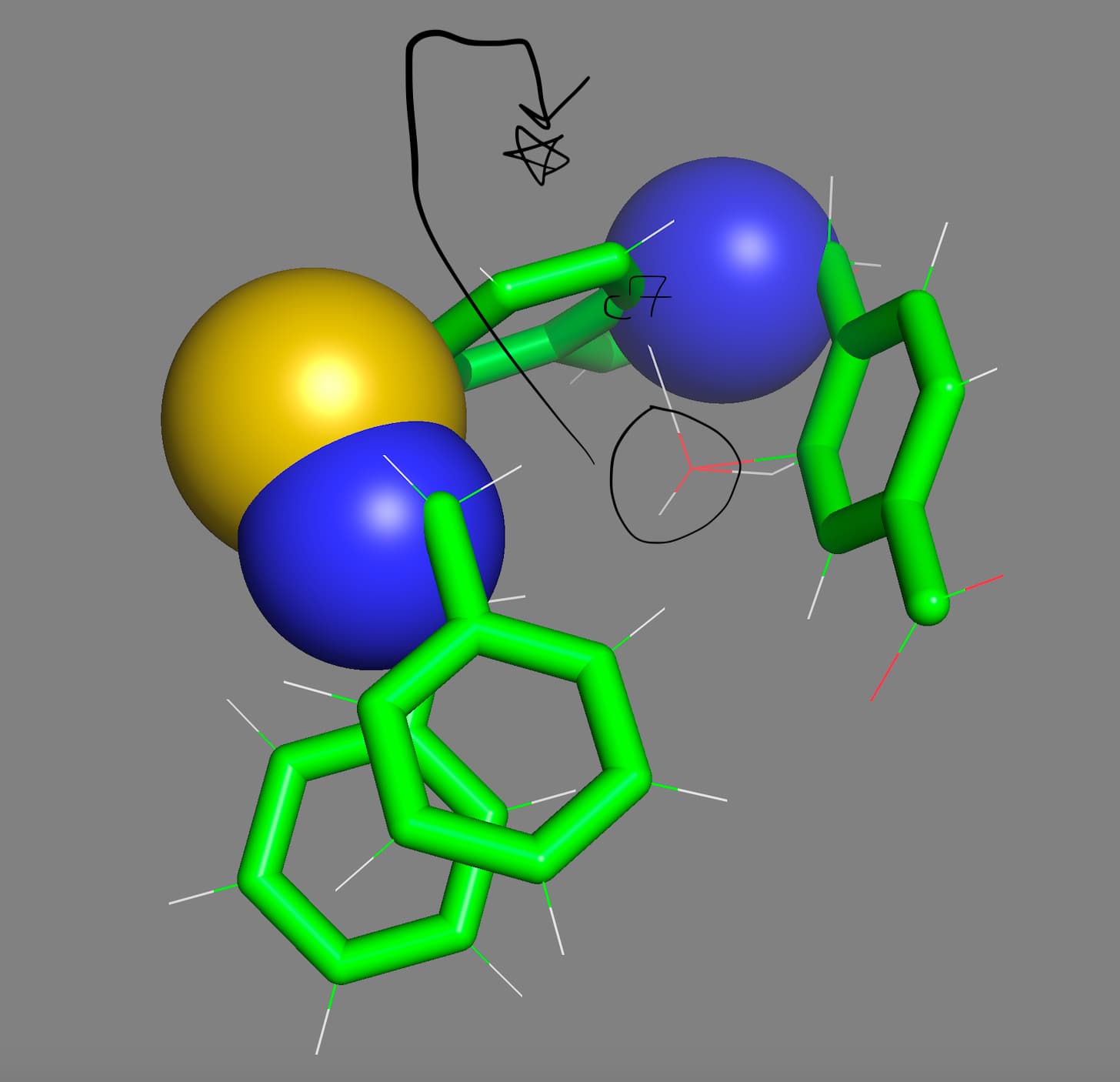
Hi ghutchis thank you for your kind update:) and sorry for me not being clear with my procedure towards what I have generated and my final aim.
I used ffTK (force field tool kit) to generate specific water cartesian coordinates that interact with the C5 Carbon via the H-atom from the water interacting with the C5 Atom as a hydrogen bond donor. All of these hydrogen bonding donors and acceptors (with just the one exhibited here) will subsequently be used again in ffTK towards Charge Optimization of the ligand to generate parametrization.
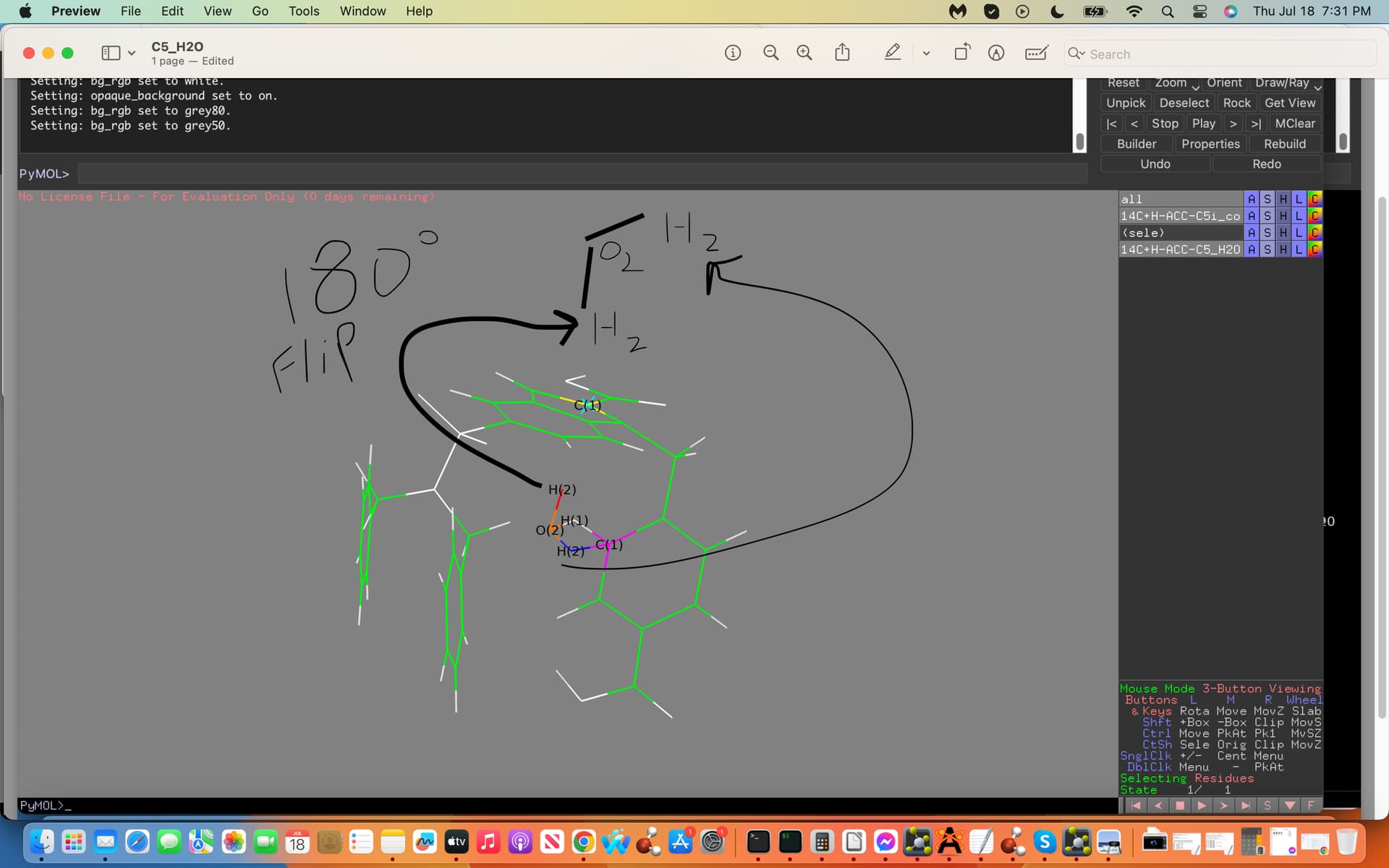
The ffTK water placement H-C5 interaction was incorrectly placed on the C5 atom allowing it to interact with an adjacent Carbon Atom incorrectly. My aim is to take the water molecule and place it 180 degrees on the opposite side of the C5 Atom, to do this I would need the `exact´ Cartesian Coordinates on this opposite side of the C5 Atom.
Attached is an illustration towards the above exhibiting the C5 in yellow with a turquois X, the adjacent C Atom in Magneta, the interacting H2O labeled (2) with the Hydrogen atom acting as a hydrogen bond donor in red (note the incorrect water interactions with the adjacent Carbon Atom in magneta). Moreover note the 180 degree flip illustrated in black exhibiting the H(2) in red now acting as a hydrogen bond donor to the C5 atom on the opposite side; this is my aim where I would need these exact but 180 degree flipped new Cartesian Coordinates to generate my proper results.
Accordingly from the above would you still suggest your centroid model and or something else?
Thanks if you know and I hope this is more clear:)
Hello ghutchis thank you for your kind update:) and methodology, success!
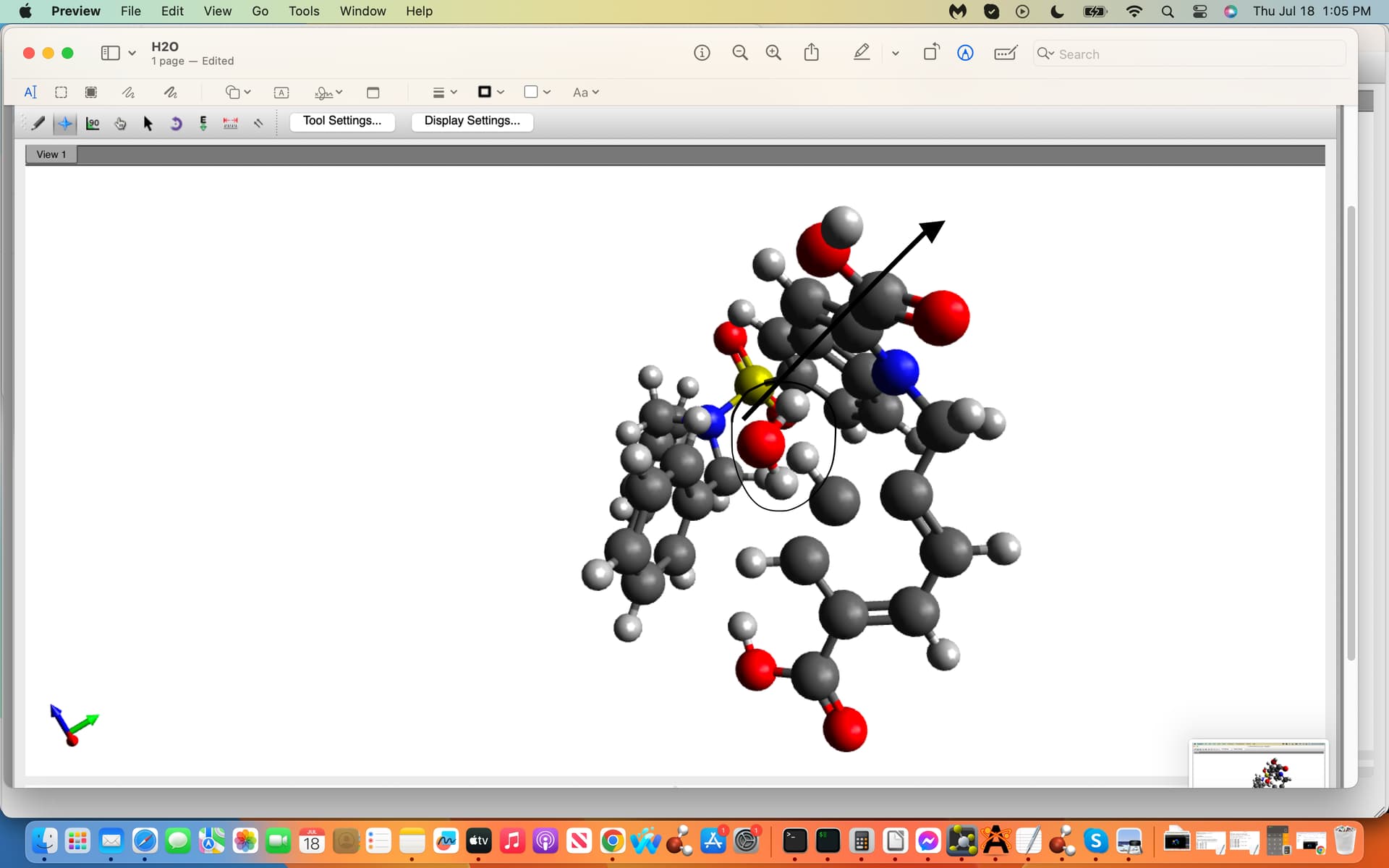
However, the water molecule I believe did not flip 180 degrees regardless
of me also trying the Rotate Around icon. Is there a way to generate this
flip as well? (See-attached
screenshots orignal, 1st and final clicks.)
Thanks if you know:)
Sorry, I just saw this was still unread!
It’s not an easy transformation - in essence, you want to move the hydrogen atoms on that water, but to a specified inversion center.
I can probably write up a Python script plugin to do it if you still need that.
Hi ghutchis no problem and thank you for your kind update that would be great if you can:),
feel free to update me accordingly.
Hello ghutchis I hope your well:) and has there been any resolution on your end towards flipping the water coordinates? (Since we chatted last I had updated the new starting coordinates before the flip, see subsequent message.) Thanks if you know:), Joel🚀
…and attached is the .xyz file exhibiting the water and C7 coordinates and a screenshot of the designated flip direction and new placement of the entire water molecule in space, thanks if you can assist:)
14C-ACC-C7.xyz (4.5 KB)
Let me whip up a script for you. (Assuming you’ve not done one already, @ghutchis? Just figured I could save you a job)
Hello matterhorn103 thank you for your kind update:) and yes that would be great since I have not been able to do this, dont know how and so far no response from ghutchis, the water should be the last three coordinates with C7 attached in the .xyz file as well, feel free to inquire if you have questions, thanks again:), Joel
This should do what you want:
invert.py (4.3 KB)
You choose the atom that should be the centre, then all currently selected atoms are inverted through that centre.
You can choose whether to just move the atoms, or leave the atoms in place and create copies (which is what you will want).
Any bonds between atoms in the selection should also be copied too.
Note you will need to know the index of the atom you wish to use as the inversion centre; see this by turning on Labels under Display Types, and under Atom Label choosing Index.
To use the script, put it in ~/Library/Application Support/OpenChemistry/Avogadro/commands and start Avogadro. The command can be found under Build > Invert Through Centre. If you don’t have Python on your machine, you’ll need to get it from python.org.
For your example, the result looks like:
where 68/69/70 are the atoms of the new water molecule.
Hello matterhorn103 thank you for your kind and detailed update:) and I will continue to resolve this and if I have any questions I will update you accordingly, thanks again:) Joel🚀
Hi matterhorn thank you again:) and I tried to generate this flip via you instructions, however,
just due to a lack of experience I am struggling i.e. I dowloaded the latest version Python 3.13 and already have Avogadro 1.2.0 installed and functioning, I believe that I would then copy and paste:
~/Library/Application Support/OpenChemistry/Avogadro/commands
into my iTerm to begin but this has not been successful, I also tried to paste this after dragging in the Python to the Iterm without success. If you have time to message specific instructions that would be great and in the meantime I will try to get savvy of how to get this code generating on my own since your time may be limited, thanks:)
You don’t need to do anything with the terminal! You don’t need to open the folder in the terminal, or run the script with Python yourself, or anything.
What you do need to do is get Avogadro 2. You need v2 to use the Python plugins. Avogadro 1 is no longer supported.
In fact, the easiest way to install the script is just to download it to your Mac, open Avogadro 2, then click and drag the script file from Finder into the Avogadro window and release. The script should be automatically installed, then after a restart of the program the menu option should be available for use. If you get asked what kind of script it is, choose Command.
If that doesn’t work for whatever reason, you can do the manual method I talked about:
Just create this folder in Finder, then put the file I uploaded into it. But if you do the drag-and-drop method, Avogadro should take care of this for you.
Hello matterhorn103 thank you for your kind, detailed and prompt update:).
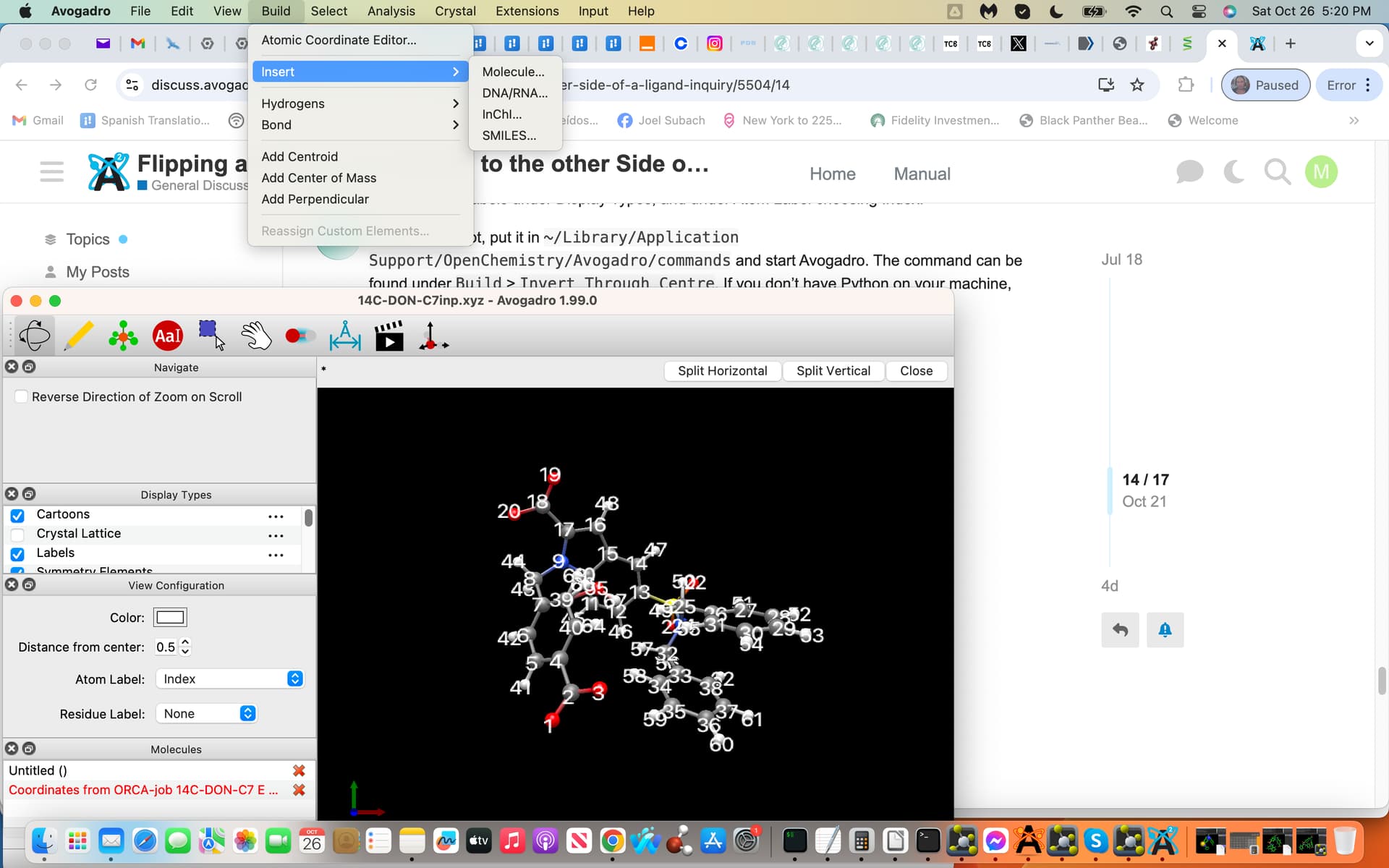
Yes I downloaded Avogadro2 and the latest Python 3.13 and dragged in the invert.py file and chose command. Subsequently I loaded my .xyz coordinate file into Avogadro2 by simply dragging the .xyz file to the screen and then releasing and subsequently labeled and indexed.
I am struggling with what I believe to be the subsequent step(s) simply because of lack of experience i.e. I could not discover this Build > Invert > Through Centre step i.e. see screenshot attached exhibiting all of the above.
Maybe I would need to activate Python within Avogadro2 and or something else, I simply just do not know? If you have time feel free to reply and if these steps should already be known by Users understandable, sorry to take up your time and I will continue to try and resolve the next steps, thanks:), Joel
After dragging the script, did you restart Avogadro? At the moment, Avogadro2 doesn’t reload scripts when you install them. (It’s high on the TODO list.)
Try restarting Avogadro. If that’s not enough, yes, you’ll need to tell Avogadro how to find the Python you installed (e.g., through Extensions ⇒ Python Settings…)
Hello ghutchis thank you for your kind update:).
Sorry I had forgot to mention in my last thread that I had only selected Avogadro → Quit since there was no option to select Restart from the Avogadro Tab.
This second time I opened Avogadro2, dragged in the .py File, released it and clicked Command but could only see the Avogadro → Quit Tab as the former. I also then repeated this same exact second process but subsequently entered Extension → Python Settings and clicked OK to Python 3.13, however again could not discover Restart only Quit Avogadro as last time. I believe I need to discover and click a Restart´ not just a Quit (I tired just Quit but when restarting Avogadro2 I could not discover this Build -> Invert Through Center similar to last time. Accordingly must I click the word Restart´? (I already tired an entire laptop restart but still could not discover the Build → Invert conduit.)
Thanks if you know:)