The next release will have previews for “Insert Fragment” and “Import Crystal”
I’m curious to get some feedback on the crystal previews, which are generated from screenshots. Right now, “fill unit cell” does not add atoms to the top / right sides of the unit cell.
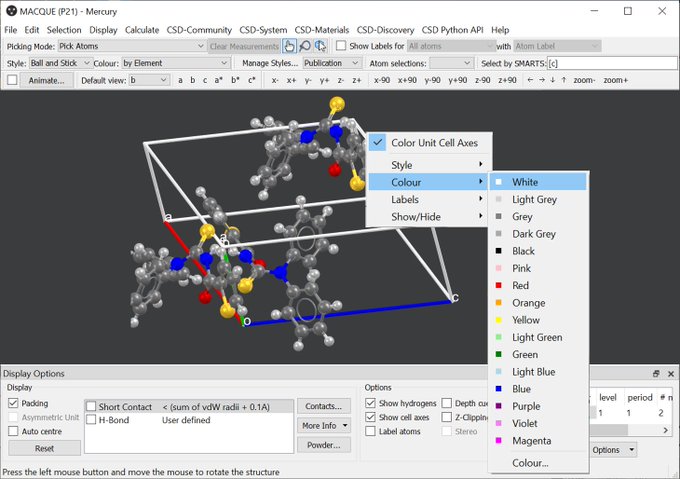
What is the perspective here? Though I don’t expect Avogadro to become a second CCDC Mercury, I do like the conventional color code along the lattice vectors a (red), b (green), c (blue) from origin of the coordinate system / the parallelepiped:
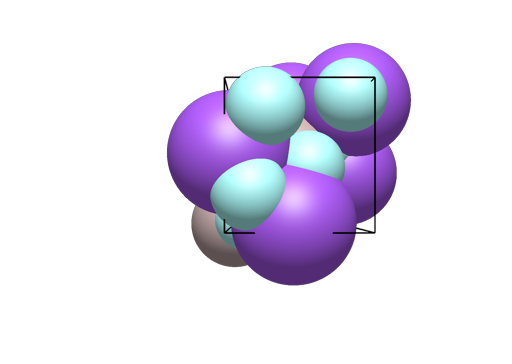
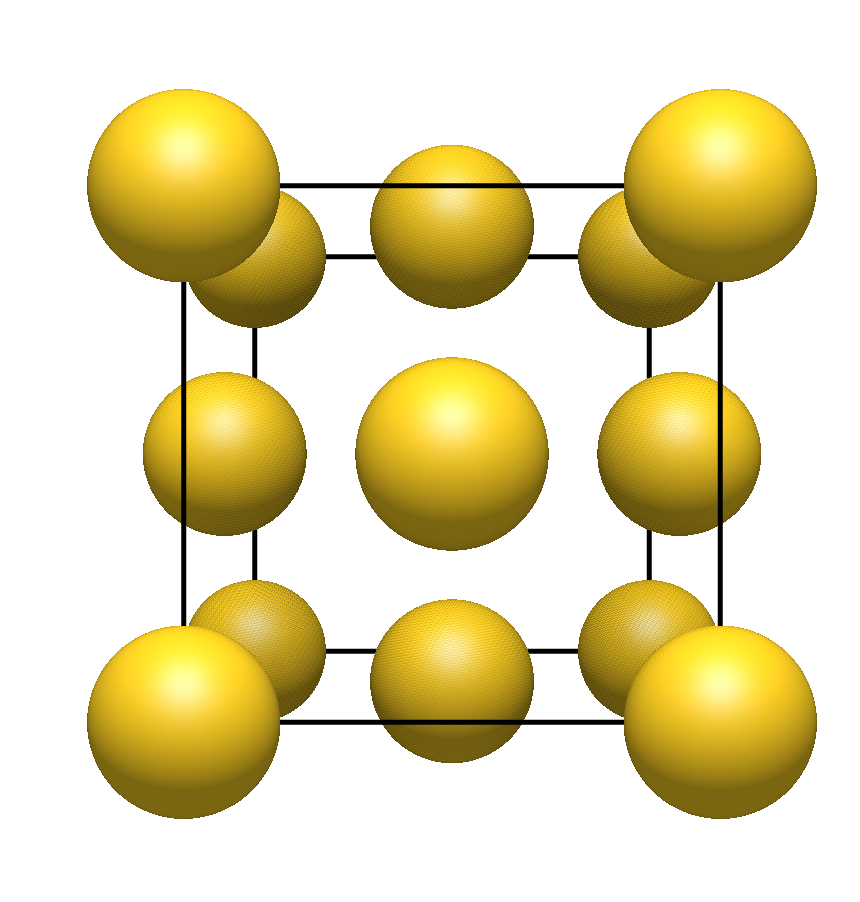
In the example of gold, there is something odd, because Wikipedia’s property box about gold reports face centered cubic as one prominent crystal structure of this metal. Scaling of the atomic radii vs the unit cell / lattice vectors is not good as atoms (or portions of the atoms) are missing. for fcc there should be one atom (or an octant, if one clips them) in each apex (sub total 1) and each side should have one (or half atom, if clipped for a sub total of 3) for a total of Z = 4:
Maybe the packing algorithm or/and consideration of the model symmetry in Avogadro is incomplete which however is pure speculation (it is more likely for me to read .cif in other programs).
It has a bit of load "9008463.cif" {1 1 1}; in Jmol to display only these four positions (space group symmetry will populate the other positions shown in the illustration of capped atoms):
I think the trick, as you’re indicating is exactly what I thought … particularly for these previews, it should populate all the positions, so people see an “expected” FCC lattice, etc.
And rotate the axes slightly.
The good news is that the automation features are useful - I can zip through directories of CIF and generate screenshots easily.
What can be an obstacle in reading a unit cell’s packing is lack of visual cue (i.e. in absence of the guiding parallelepiped) if the projection is in parallel/orthographic view, or in perspective. Both approaches have their place, they complement each other.
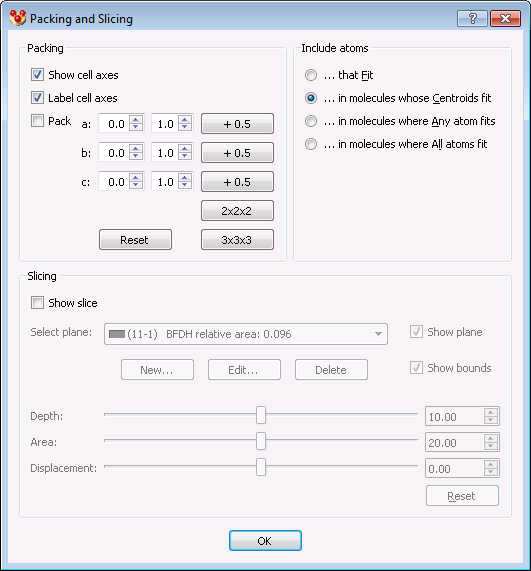
Referring to packing to fill the unit cell, the corresponding widget in CCDC’s Mercury provides these defaults:
The idea to zip through the .cif [as an example of a structure file format] within a folder is an idea very welcome as it could close a gap perceived because (aside from the file preview by e.g., ChemDoodle, or Marvin) thumbfish to best of my knowledge still is Windows-only.
I probably won’t have a chance to get all those features in Mercury anytime soon - they’re great and I’ve definitely used them, especially for generating molecular clusters.
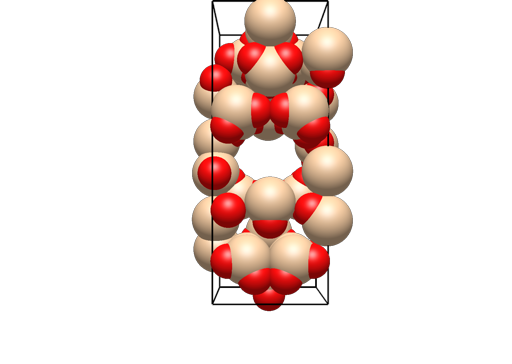
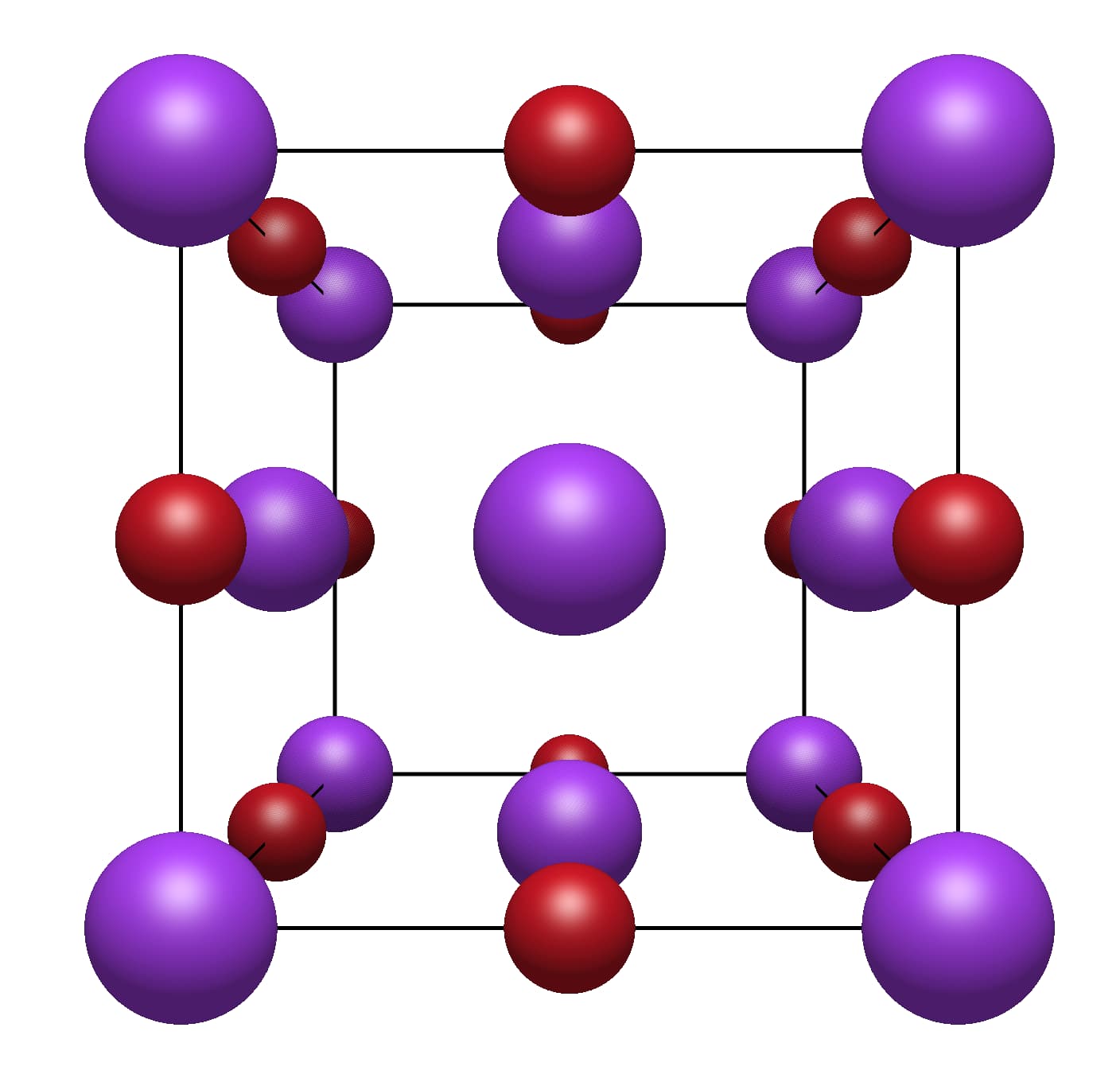
What I did manage, is generating all the copies on the unit cell boundaries, e.g. KBr
The illustrations are accessible because they don’t appear overly crowded. Perspective and change in tone are in sync to convey space, too.
Regarding the three choices, I personally would go for the symmetry unique atoms (i.e., only one gold atom) as default however equally recognize this can be hurdle for the untrained eye if i) at one point equally applied to molecular crystals if ii) a molecule is on a symmetry operator of the unit cell symmetry (e.g., centre of inversion, mirror plane, axis of rotation). Because then, the molecule appears to be a fragment while (by the data in other blocks of the .cif file) it already is completely described. To borrow a term from Mercury’s manual, if there is a difference between the asymmetric unit and the crystal chemical unit. (For the purpose of illustration, I attach an example of benzene, where the asymmetric unit consists of a C3H3 fragment eventually “completed on the fly” to C6H6 by Mercury.)
I speculate the display of the content of the asymmetric unit is implemented easier, than the one of crystal chemical unit. However because Avogadro addresses an audience with little to none background in crystallography would let the display of the crystal chemical unit to be the default with a toggle in a side pane to get the asymmetric unit only.
Additional thought: some of the sources COD uses are less (organic) molecule oriented, but mineralogy/geology. It could be worth to get in touch with them.