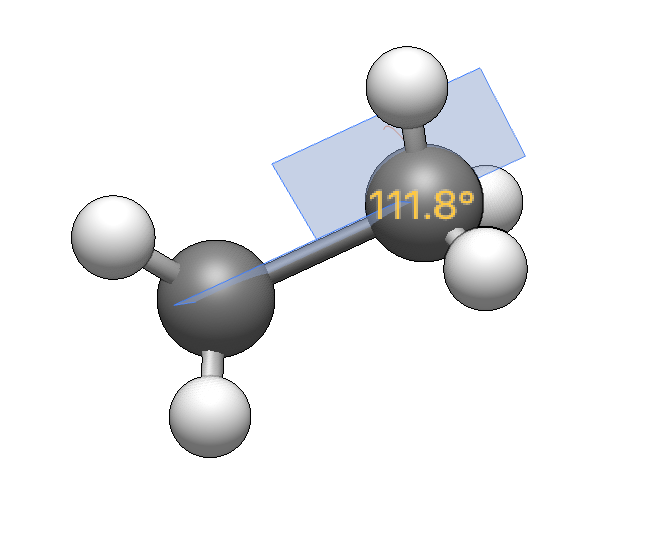
Other editors like GaussView have a very nice feature which lets you select up to four atoms and modify the associated bond property in a popup window using a slider and/or numerical input field. Since I haven’t found a way to do this in Avogadro2, this would be a great addition to the measurement tool.
If you select two (not necessarily bonded) atoms, and right click / shift click, etc, a popup window should appear with which you are able to adjust the atomic distance interactively. The popup consists of two drop down menues, a slider, a numerical input field, and an OK / Cancel button pair. The left drop down menu is for the 1st atom(ic fragment), the right drop down for the 2nd atom(ic fragment). Both drop down lists consist of the entries “fix atom / fragment”, “move atom” and “move fragment”. If you change the slider (default range 0.5 to 5 Angstrom, initially set to the current atomic distance) or enter a numerical value, the two fragments are interactively moved apart to the specified distance, obeying the settings in the drop down menus. (If both parts are set to move, they move symmetrically.) Once OK is clicked, the popup closes and the new coordinates are applied permanently. If Cancel is clicked, changes are discarded and the previous molecular coordinates are restored.
Analogously, if you select three atoms, a nearly identical dialogue appears in which you can interactively change the angle between the (not neccessarily bonded) three atoms. Which of the fragments move is determined by the drop down menus as before. Naturally, the slider defaults to a range of 0° to 180°, initially set to the value of the current angle.
Similarly, if you select four atoms, the dihedral / torsional angle can be interactively modified. Atoms 2 and 3 specify the atomic axis to rotate around. Which atoms move is again specified by the same drop down menus. The slider defaults to a range of -180° to 180° and is set to the initial value.
@ghutchis BTW: Happy birthday.