Avogadro version: 1.99.0-137-gf875369
Operating system and version: Windows10
Expected Behavior
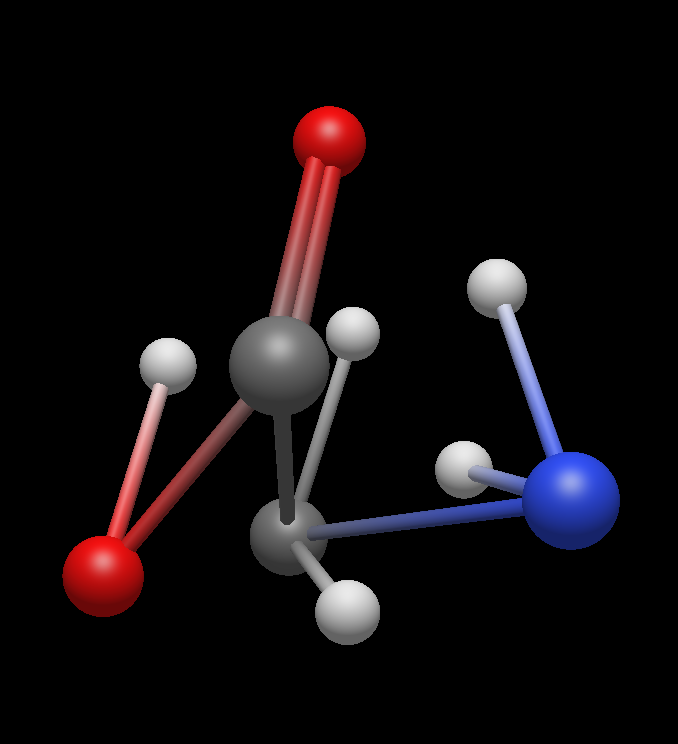
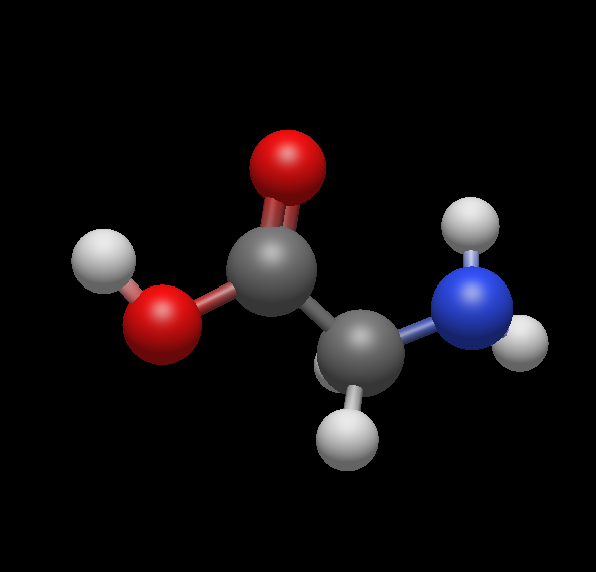
Standard molecules available from the Build → Insert menu change their geometry under Extensions → Optimize remaining recognisable
Actual Behavior
The geometry changes completely and the result does not make sense. Further investigation shows that the LJ force field is used. When attempting to switch to another force field, no optimization takes place at all. Is it related to the inability to download plugins from the Extensions menu?
Do you have Python 3 installed on your computer? The force fields beyond LJ are Python scripts for now, until we can finish a native implementation of something like UFF in C++.
Yes, definitely. For example, I do not experience problems with xTB optimisation that, as far as I understand, also uses python. I also did not have a problem with the force field until I changed Avogadro2 to the latest version.
Welp, I have downloaded and double checked the problem on Windows 11, and it seems that it is a bug in Avogadro. I’ll take a peek at the latest commits and try to pin things down as best I can.
In the meantime, using the OpenBabel implementation still works, so I would recommend that for any force field things. Hopefully we can get this sorted out quickly!
Yes, OpenBabel does ineed work, thanks! I am not sure, however, what am I actually doing by using this option - I mean what kind of force field is used in the OpenBabel optimisation?
Also, prior to optimisation, I am building a peptide from amino acid molecules that are available. It would help me a lot if I could use one amino acid and manipulate (translate, rotate) it independently of another. This should be done via Select, but I do not see an intuitive select option where I could click on a number of atoms and make them my selection for the geometrical manipulation. I also do not see at intuitive option to select one atom and change its position distorting its bonds. The manual seems to be talking about Avogadro1, so not of too much help… I am sure I am missing something extremely basic!
Yes, thanks a lot, I was embarrassingly just a click away from discovering that:) And changing the force field does change the geometry slightly so it seems to work!
Once you have some atoms selected, you can use the Manipulation Tool on the whole selection at once. The link covers this scenario and describes the behaviour. I did the recent rewrite of the tools docs so if anything is unclear, let me know!
As in just a normal movement of a single atom while keeping everything else in place? That’s just simple click and drag with the Manipulation Tool when no atoms are selected.
Thanks a lot, very clear and helpful! My main mistake was that after selecting one molecule, I was trying to rotate it using the Navigation tool, while what I needed was the Manipulation tool, so I thought my selection did not work… Anyhow, life is quite a bit better now:)
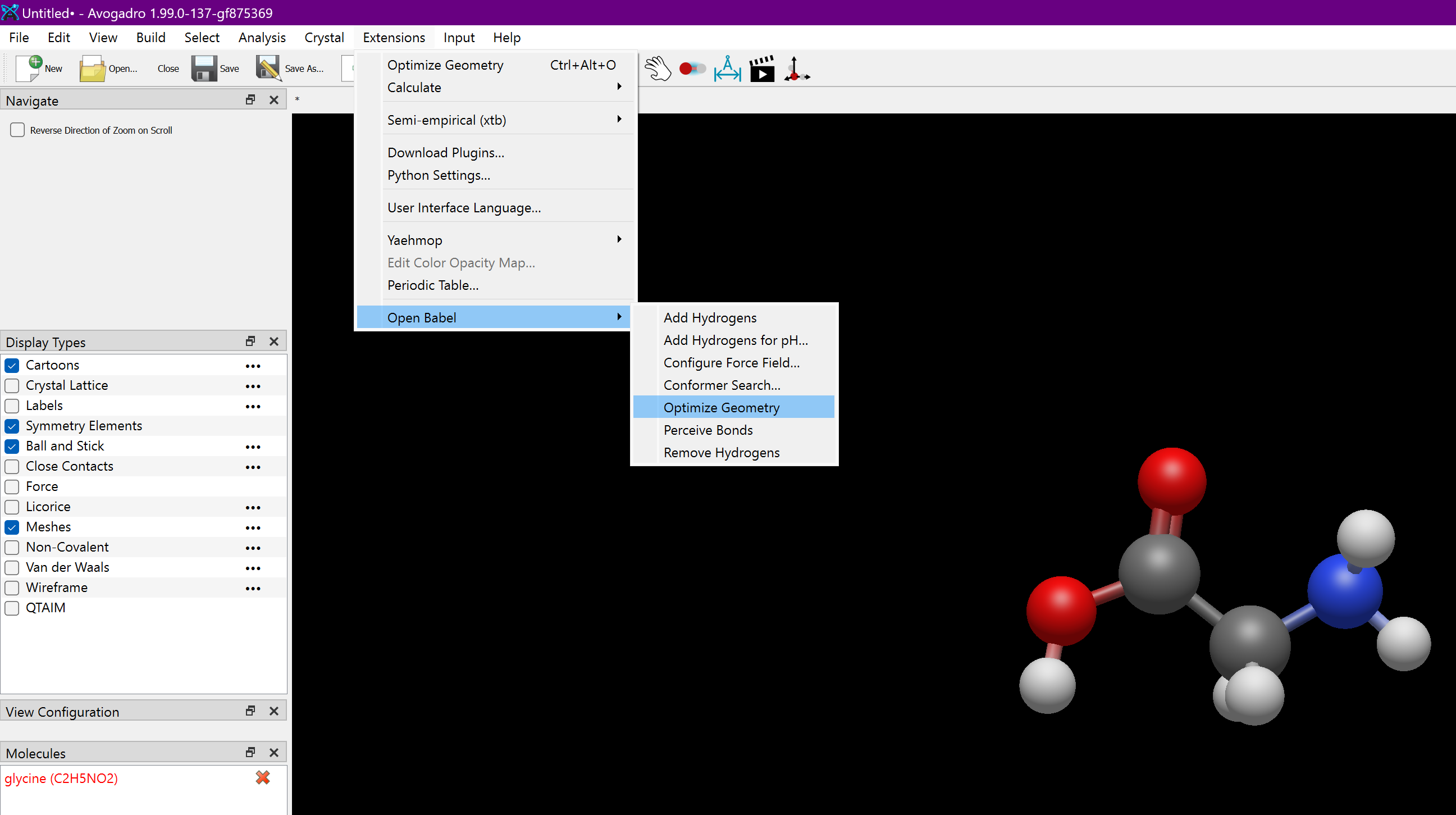
No, not quite. The default builds on Mac and Windows should have integrated calls to the Open Babel force fields as well (UFF, GAFF, MMFF94). The auto-select process is to try GAFF first, then MMFF94 and then UFF as a fall-back.
If the only force field available is LJ, that means that Avogadro was unable to load the Open Babel implementation internally. (The separate menu option calls obabel as a separate process.)
Thanks a lot, @ghutchis! By “plugin”, do you mean it would work as a plugin for Avogadro? Do you plan to include it into the list of plugins downloadable from the Extensions menu? Thank you!
I think I might be a bit confused. I can see in an old installation of Avogadro 2 that there is a folder at C:\Program Files\Avogadro2\lib\avogadro2\scripts\energy that contains 7 files:
ani2x.py
gaff.py
gfn1.py
gfn2.py
gfnff.py
mmff94.py
uff.py
In the most recent installation of Avogadro 2 that I have, a nightly build downloaded today, the gaff.py, mmff94.py, and uff.py files are missing. I tried just moving uff.py into the new version’s folder, and it didn’t work. I always assumed that the various python files there were the way that Avogadro 2 ran force field optimizations, but is that not correct?
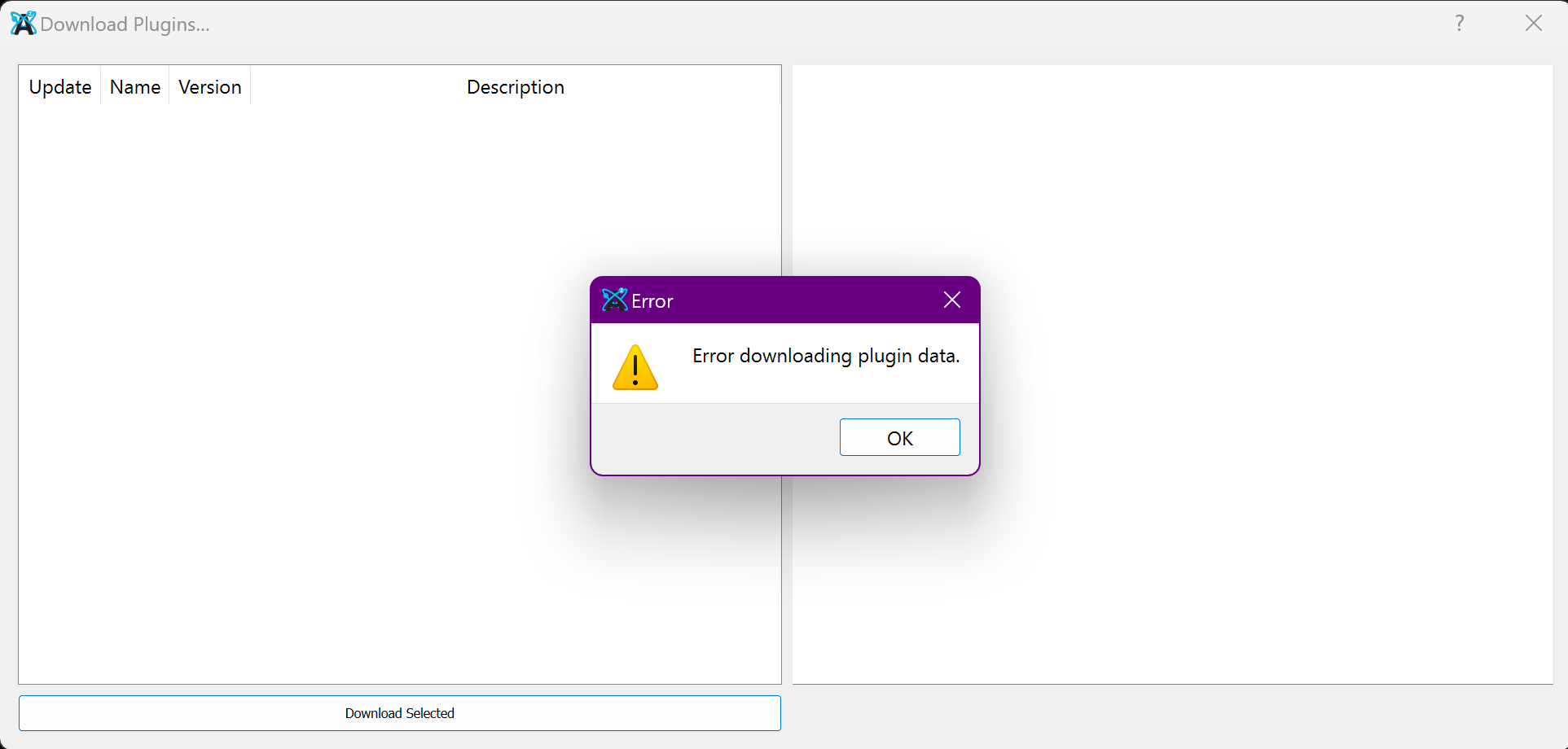
On perhaps a related note, the plugin download menu does not function, and shows the error here:
I cloned Avogadro 2 fresh today from the git page and built it myself, and my version has a functioning plugin download menu, however the force fields still don’t work. I’m not 100% sure if any of this is helpful, but I figure any information is perhaps useful information.
That’s great, many thanks to @Dhruv_J! However, I cannot download plugins under windows at the moment, so I will probably have to wait until this issue is fixed. Would be grateful if you could announce once this functionality is restored for the windows users.
I believe the issue they are experiencing is that UFF, MMFF94, and GAFF are shown as options in the drop-down under Extensions > Calculate > Configure but that when they are selected as the force field, Extensions > Optimize does nothing.
If this is recent, I think it might be caused by the “deferred start” I added to the force field code - registering these a bit late so the window appears quickly.
I’m currently building this - please let me know if it fixes the problem:
@avervit - have you had a chance to check a new nightly build? Does the geometry optimization now show UFF, etc and optimize OK without using Lennard-Jones?
Trying to get an idea on which bugs are fixed for a 1.100 release soon.
Sorry, I checked the new nightly build a few days ago but only for the plugin download feature and it did work - I commented on that in another thread. However, I see now that the geometry optimisation is still possible only with LJ force field. So, this one has not been fixed. The version I am looking at is 1.99.0-148-gd4a1e87, downloaded on the evening of December 22nd US time. If changes were supposed to occur since then, I can try the more recent version.