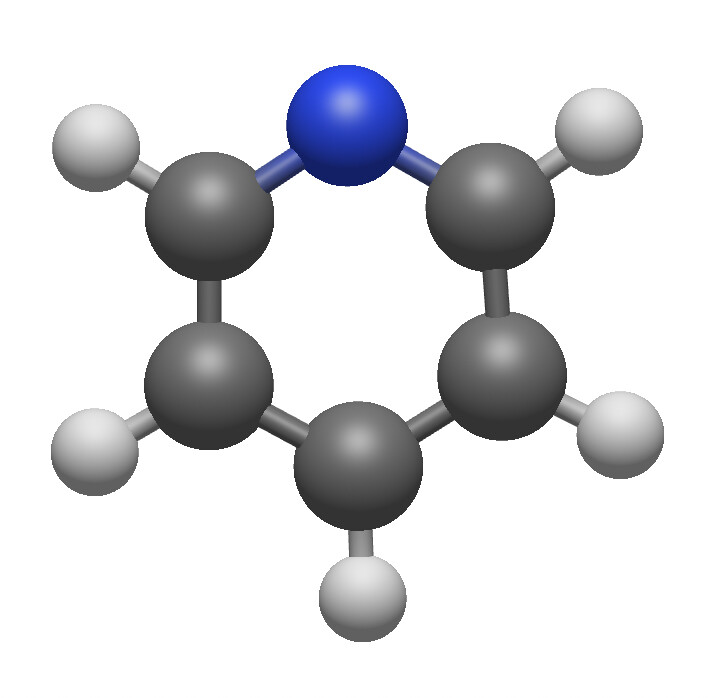
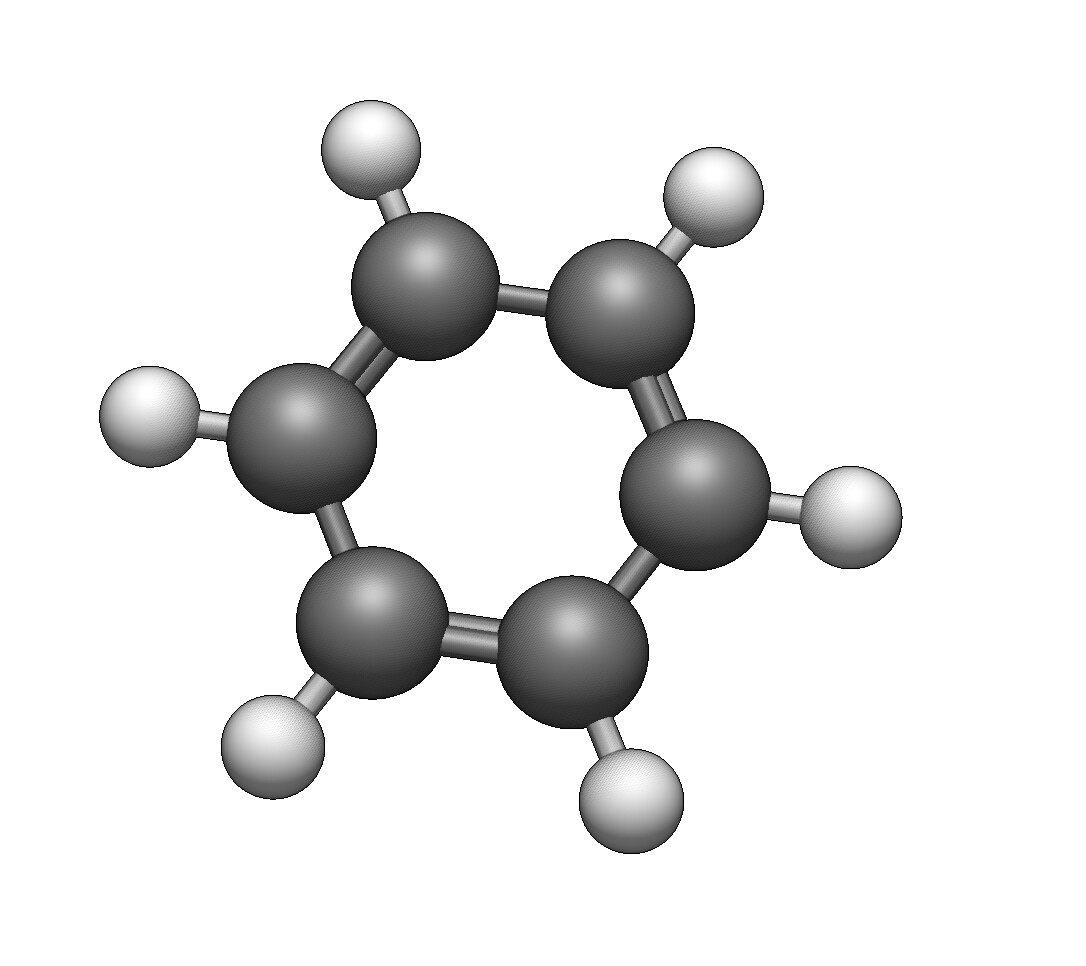
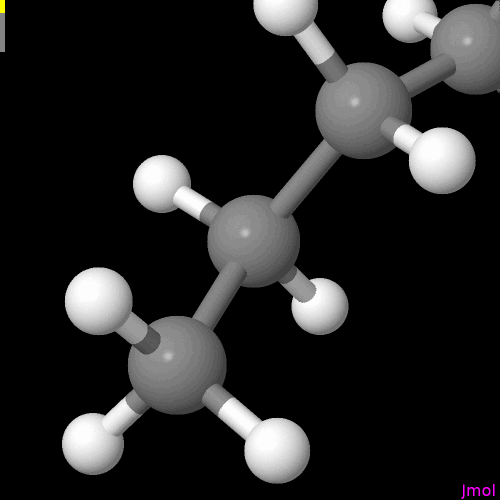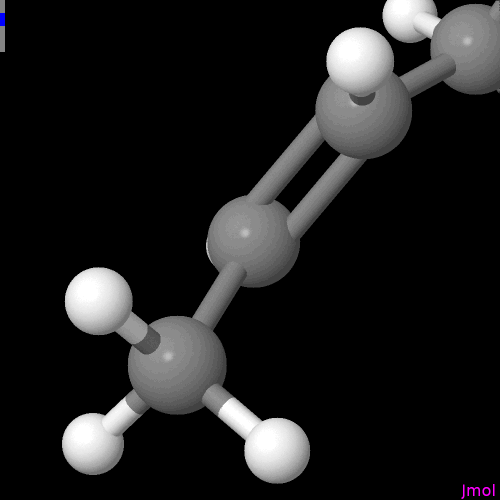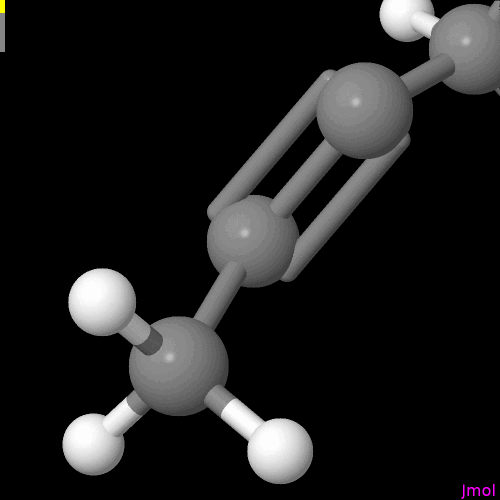
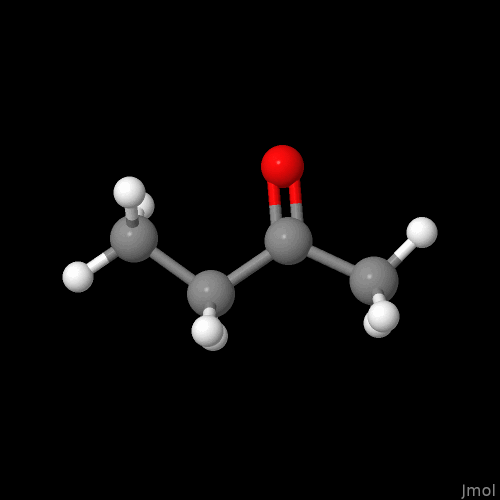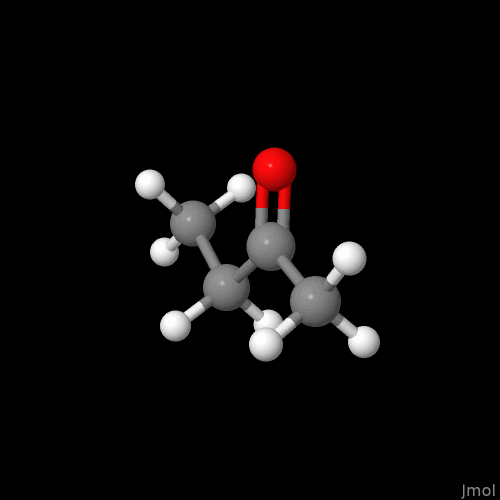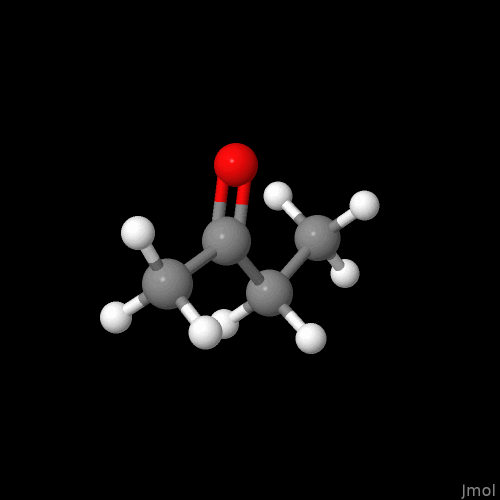
I’m not sure if this is intentional or on the radar, but the double bonds are displayed so that you can only see them if you look at a single plane (my best guess - xy plane).
C -0.037422 -1.249404 -0.208981
C -0.010729 -0.346733 -1.260140
N -0.026235 -0.887743 1.092553
H 0.021681 0.702030 2.405624
H 0.073898 2.465057 0.636840
C 0.013465 0.437359 1.352252
H -0.021403 -0.693862 -2.286993
H -0.069480 -2.319671 -0.391212
C 0.029959 1.011006 -0.960693
C 0.042415 1.414813 0.370310
H 0.051779 1.748271 -1.757965
The problem arose when I built one molecule, removed all atoms, and started creating a new one, and apparently, I started doing that while in a different plane. Is there a reason why the space doesn’t reset to a top view on an xy plane once you remove all atoms?
Generally, I go by the “principal of least surprise.” If you rotate the camera, then delete some atoms, why should the camera change?
I think the right solution is to make sure the code picks a direction orthogonal to the bond, but “diagonal” to the XY plane. I think you’re right that when it generates the orthogonal displacement, it’s always in the XY plane.
So we want to take some of that XY displacement and put it into Z as well.
I agree; I meant you wouldn’t notice if it changed if you removed all atoms. I thought this would be an issue only in such cases of resetting everything to 0, but now that I think about it, there may be other cases.
The question includes where the bonds are located, their diameter (in the representation as cylinders) vs how far they are spread apart of each other when connecting the atoms. E.g., Jmol
For the purpose of demonstration the molecule was neither moved, nor submitted to (re)run an geometry optimization after reaching out for the alkene, and alkyne. I know the overall conformations shown are problematic.
However yes, Jmol equally identified a clever implementation of the problem because no matter the perspective you have on the C=O of e.g., MEK (load $methylethylketone;) one of the “two cylinders” does not obstruct the view on the other one: