I use Avogadro (the modified for ORCA version to be precise) a lot, both for computational chemistry and visualization. I really love it and in general it does everything that I need very well, but recently I have started working with big aromatic systems featuring very delocalized bonds ; while for the comp-chem side it does not change anything, I was wondering if there was a way/plugin to display delocalized bonds (dotted-lines or similar) for visualization.
In any case, thank you for the great work (and have a nice weekend)
Potentially. I think the first question is always “what would we want it to look like?” Defining the use case and the desired look / visualization … even if it’s a rough draft makes it a lot easier.
So are you looking for “1.5 bond” dashes in a benzene, or do you have something else in mind?
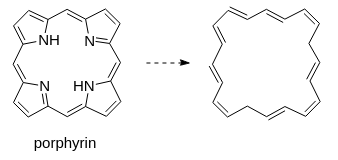
So, it would probably be something like that indeed, but possible to expand outside a “traditional” five/six-atoms aromatic ring. I am working with porphyrins, which have this very characteristic massive aromatic ring, and since it’s at the core of why I am working with them, I would really like to be able to display it a bit more “properly”, if that makes sense.
For this reason, what would really be best would be a “1.5” bond option with a single bond coupled with a dashed one of around the same thickness, instead of the kind of (sometimes dashed) circle people sometimes draw inside simple aromatic cycles.
EDIT: of course I would not expect such bonds to be compatible with the Molecular Mechanics modules, it would be purely for visualization on an already optimized structure.
I’m not sure what you refer to with “this very characteristic massive aromatic ring”. If simply trim off the inner of ChemDraw’s template about porphyrin (no peripheral decoration; no Zn, Cu or other metal complexation), aromaticity is absent:
Regarding the highlight of interesting sub structures, I just become aware that a SMILES based substructure search and highlight option is available in Jmol.
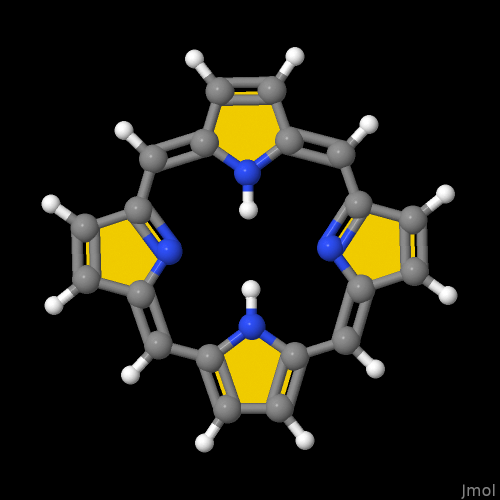
Because this thread is about porphyrine, for example, the instruction to the the program’s console are
to obtain a conformer from NIH’ servers followed by the user defined highlight of pyrrole structures which looks a little bit like the custom of colored rings by KC Nicolau & co-authors in papers and classics in total synthesis
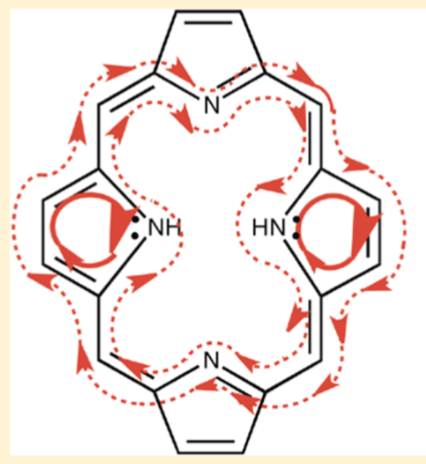
So, this is more or less off-topic anyway, but you missed the aromaticity because your trimming basically removed it entirely. For an overview of what’s going on in systems with a porphin core, this image is a good representation:
I agree that it is a nice feature, but that is precisely the kind of things that does not really work for complex systems like porphyrins, only for more “classic” cycles.
to load the structure, dismiss all information about a bond order greater than one, and to reassign aromaticity in the visual depiction.
I don’t know if one can explicitly instruct Jmol to consider either the inner circumference, or the outer one. Depending on your intent to highlight, it might matter.
Edit: in the visual I departed from Jmol’s defaults. Now: atom size to 20% of the van der Waals radii and bond radii of 0.05 A (instead of 0.15 A).
At the moment, Avogadro2 doesn’t have any aromaticity code … intentionally.
But it would be fairly easy already to write a command script in Python that uses RDKit or Open Babel to select the appropriate atoms via SMARTS and change the atom colors or otherwise highlight them.
Come to think of it, I’ll add a “Select atoms by SMARTS” RDKit script (and parallel Pybel script) in a bit.
I would like to add SMARTS matching to Avogadro, but it’s probably something for after 2.0 with the other more pressing roadmap items.
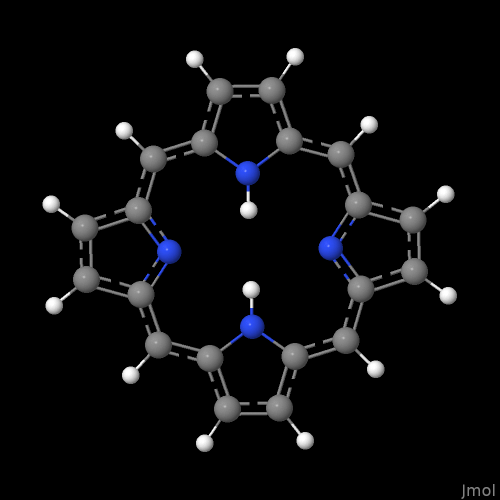
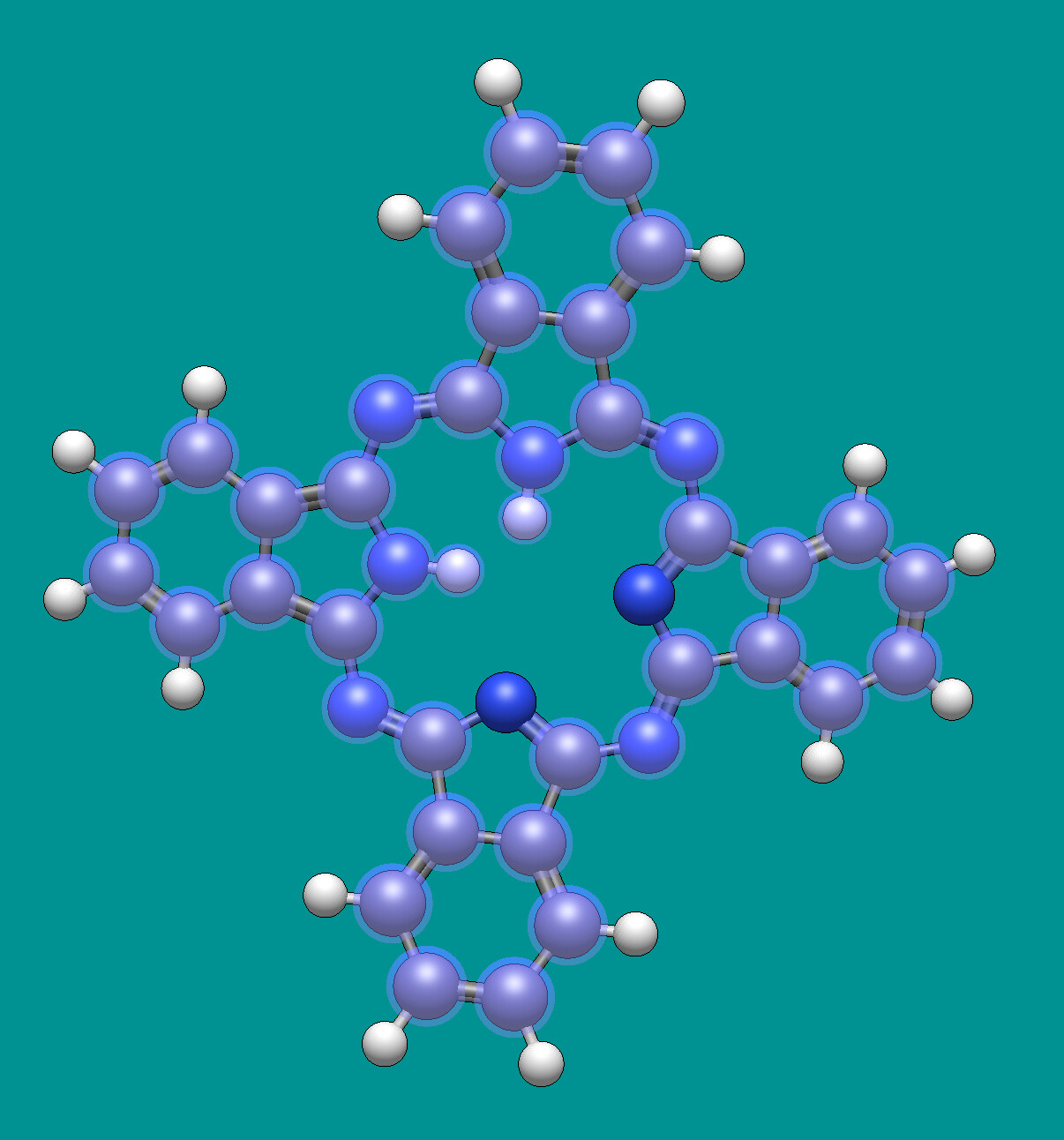
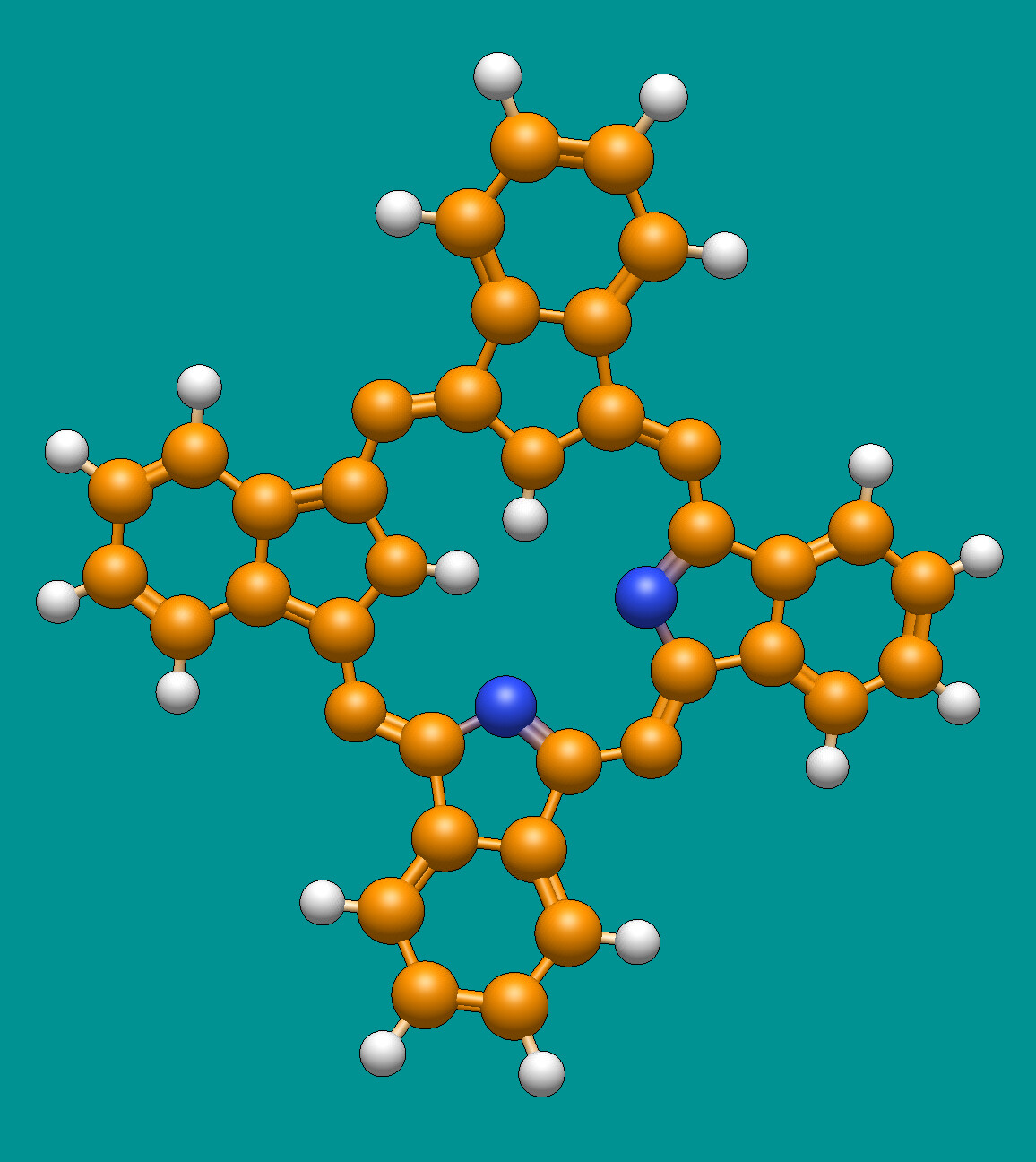
For example, I just picked “a” for aromatic atoms and it ignores appropriate nitrogens in phthalocyanine. (The protonation is weird - I used “download by name” for this example.)