Together with being able to pass multiple fileformats, it should be straightforward to implement these constraints using the appropriate functions in RDKit before minimization.
I would also like to get these functionality using the Parsley forcefield/OpenMM but the implementation seems not as straightforward.
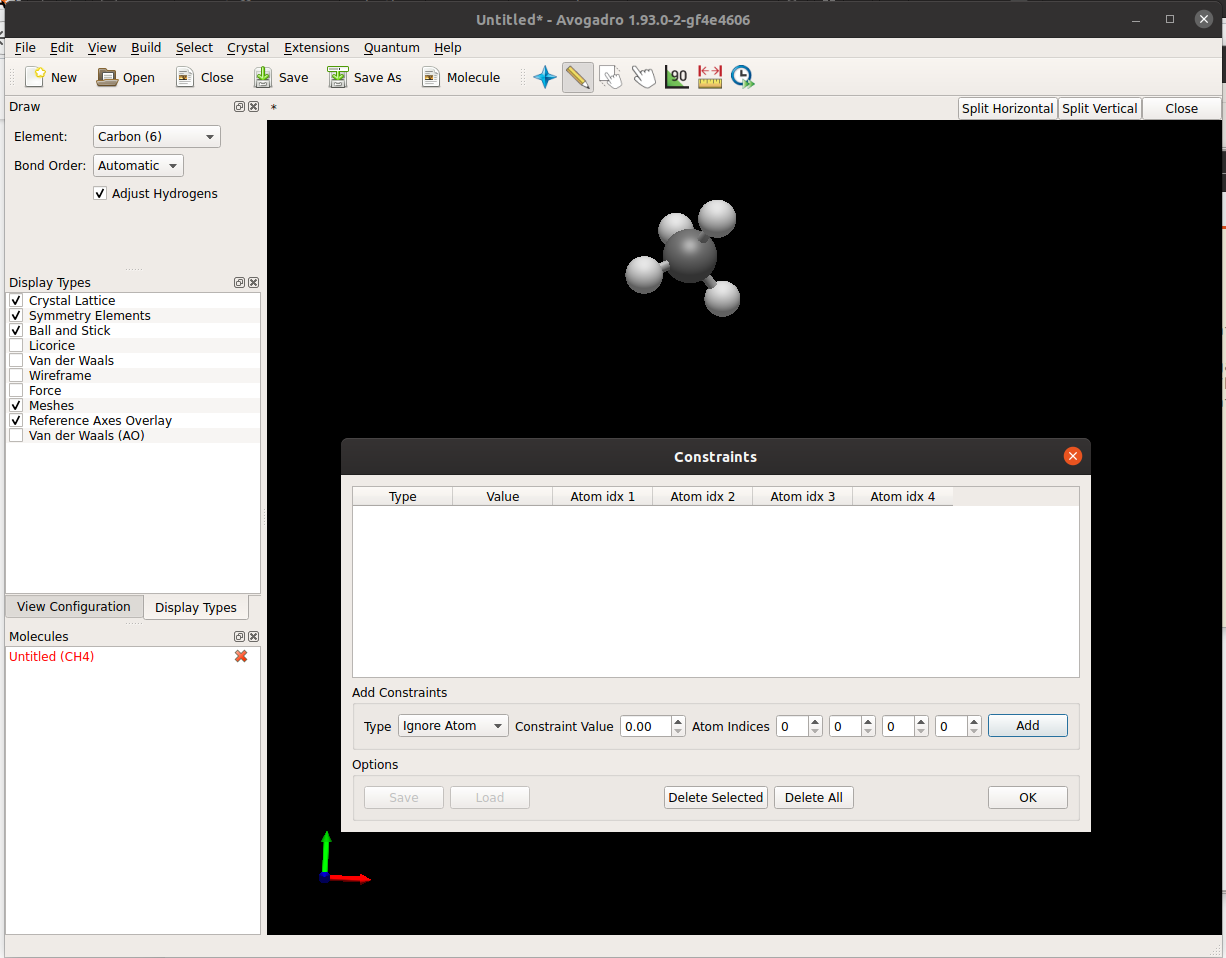
Yeah, I’m not 100% sold on the user interface. I’d like to adopt the Atom / Bond / Angle / Torsion spreadsheets from Avogadro 1 and then make it possible to add a constraint from that dialog.
Ideas welcome.
The JSON seems reasonable. The catch is:
Implementing the UI
Adding the constraint code to the new force field engine
Passing the constraints into CJSON and other sources. (commands, generators, etc.)
I can imagine it would be nice to pass the constraints to QM interfaces to enable constrained optimization in Gaussian / Orca, etc.
Keeping the spreadsheet dialog from Avogadro1 was exactly what I had in mind.
I admit that I did not think at all about the QM input generators - I agree that it would be nice to be able to apply the constraints there, too. I have only some modest experience with MOPAC, setting the optimization flags for specific coordinates is rather tedious within the input file itself (maybe I miss a trick or two).
What comes to my mind is something like ASE. That could provide a generic interface for implementing constraints but might also bring a loss in flexibility further down the line because of the calculator interfaces implemented there.
Definitely willing to do some C++ - but no experience. I would like to try but that’s something in the longer term. My knowledge of C++ is superficial to point that I was even confused by the expression “give some pointers” (in combination with an insufficient command of English ).
I added a constraints attribute to the QtGui::Molecule class definition to store the constraints - maybe there is a more “plug-inic” way of doing that ? Basically it is the code from Avogadro 1.2 adapted as a Plugin for Avogadro2. As this ismy first attempt of programming in C++ it is probably quite a mess and needs some review.
On top of that would it would also be my first contribution to a an open-source project - so, what would be the next step to proceed (pull request?)?
I am still working on an implementation of the Constraint class itself. In the beginning I had something really simply that was basically just an array of numbers
But I figured that it would be more convenient to somehow link the constraint directly to the atoms, so that changes in the molecule would automatically be reflected in the contraints. But I am having some trouble to understand how the atoms are actually stored - are there any “Atom objects” that persist and that have their characteristics updated upon molecule changes?
Your data structure is essentially what I drafted, using an enum for the constraint type (distance, angle, torsion)
Atoms are stored as indexes. That makes it a bit difficult to track constraints - there probably needs to be some code particularly to update constraints when atoms are removed.