For a molecular orbital, you’re evaluating the coefficients for each basis function on each atom. So you get something like:
\sum c_a\psi_{a}(r_a)
For the electron density, you’re essentially doing that across the electrons in each MO.
You’re talking about cube files - these are simply a format with an indication of the xyz points, the size of the “cube” (i.e., both distance in Cartesian and number of points along the xyz axes). For these, some program has already calculated the values of the molecular orbital or electron density and stored it in the “cube.”
So if you read in the cube, you’re simply just looking for the points in space which hit the specific iso value. Avogadro previously used the traditional “marching cubes” method and now uses the more efficient flying edges algorithm.
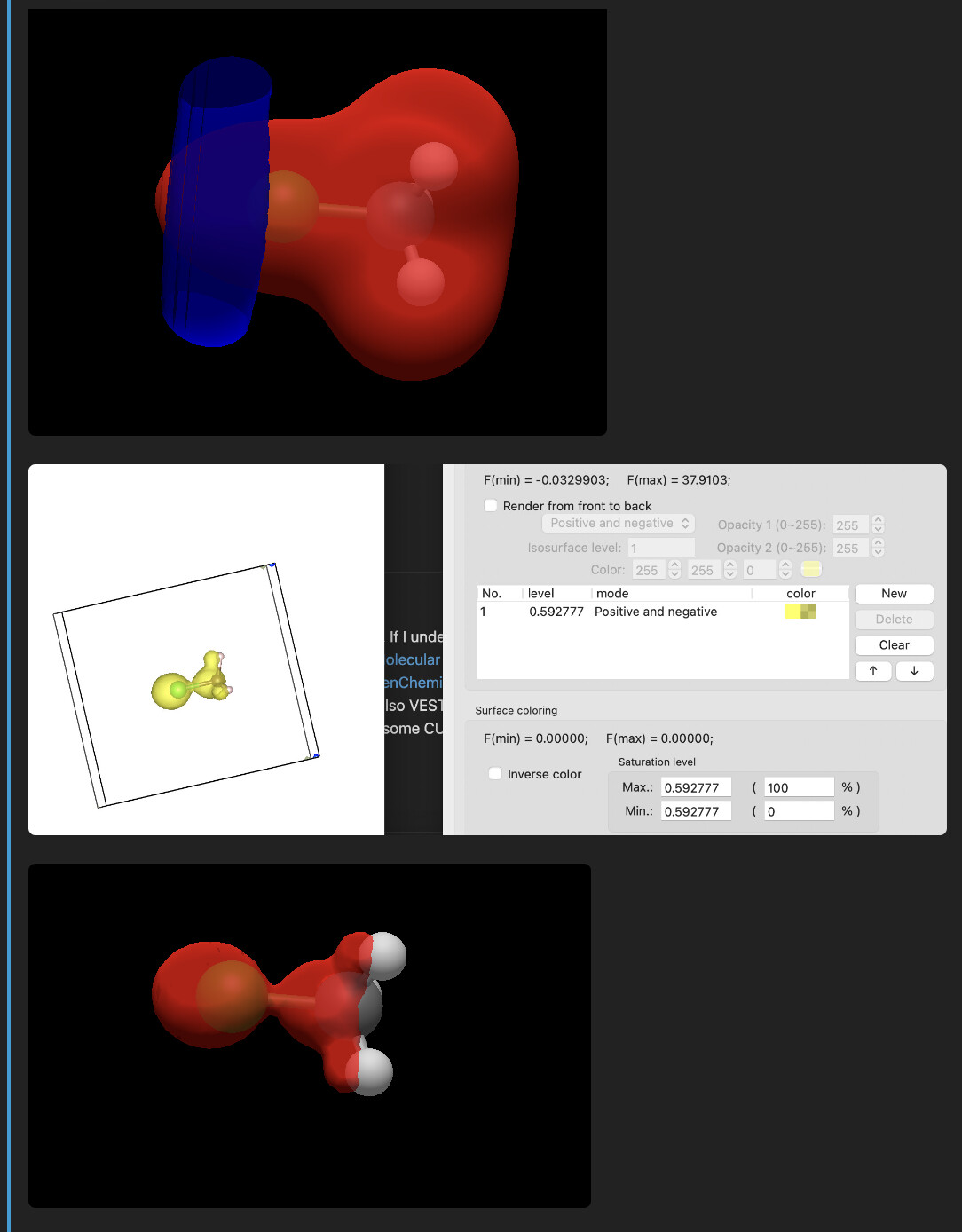
First off, 0.03 is pretty customary for molecular chemistry. But IMHO, it’s something that the user should be able to adjust. If you want a different value, you can absolutely do that easily.
I’m not at all sure what you mean by “physically more correct” from an electrostatic potential cube. That cube indeed has both positive and negative electrostatic potential, as reflected in the Avogadro rendering. Your screenshot from VESTA only shows one phase. Of course that’s due to the choice of isovalue.
How is it “correct” to only show one phase when both positive and negative electrostatic potential is present?
Yes, the iso value is adjustable, I agree. Now I understand that I was thinking about good initial values (defaults). I can see that for MOs, 0.03 usually makes sense, not sure about densities, etc.
I’ve figured that those powers in the fortecubeview code are coming from normalization.
MO (WF coeffs, as you mentioned above) with power 2 is normalized to 1. Density with power 1 is normalized to the total number of electrons. Confirmed both on some CUB files. ESP I’m not sure.
In principle, electrostatic potential should integrate to the total charge of the molecular species. For example, if you have \ce{CF3H} or \ce{H2O} the positive and negative regions should balance, because they are neutral molecules.