I’m a student trying to create some models for a research project on neurotransmitters interacting with functionalised polymers. When I save my progress and load it up however, the force field optimization and auto optimization doesn’t work at all.
The software says it’ll revert back to UFF, and that’s all it does. It just keeps reverting back to UFF every time I press ctrl+alt+o without actually working.
If somebody could please help me out with this I’d highly appreciate it. I really need this software to work for a few papers I mean to publish.
I don’t quite understand what you’re trying to do, or what molecule you’re trying to optimize.
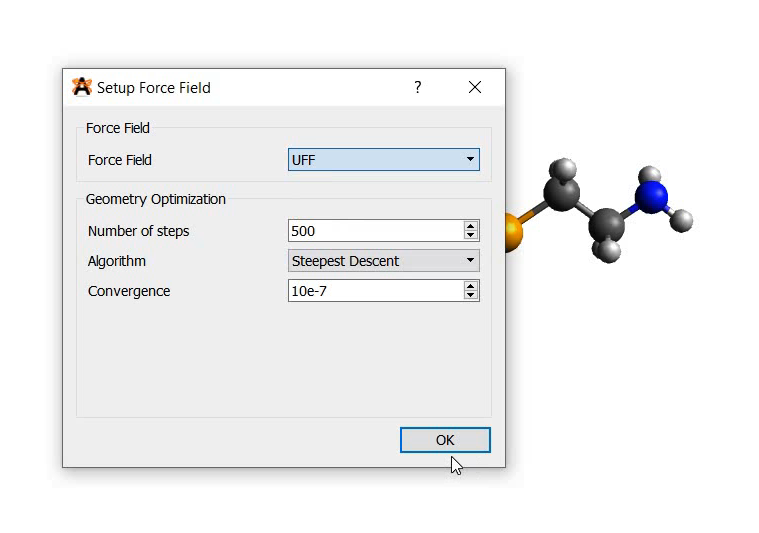
If you have the optimization set to use MMFF94 and have elements in your molecular that are not in MMFF94, it will naturally revert to UFF, a universal force field. That has nothing to do with saving or anything else - it will happen based on your molecule and the force field chosen in the options.
What’s happening is when I load up a model I have created and saved in .cml, I cannot apply any force field optimizations to it, be it MMF94 or UFF, whereas the model was optimized in MMF94 before saving.
Hi,
I am a new user and I encountered the same problem.
I built a model and I successfully optimized the geometry the first time.
Then I copied and pasted the model in a new file and I modified it by adding a new element.
I tried to launch the optimization but it failed and the message “cannot set the force field” appeared.
Please could you tell me how can I fix this issue?
It would help to know what the new element is, or have a link to the file. For example, the MMFF94 force field supports common organic elements, but not a very large subset (e.g., B or Se are not supported).
The element is sodium. First I optimized the EDTA geometry then I replaced one proton by a sodium and launched the UFF optimization but it did not work. What is the trick?
To be more specific, when I import or build a model, the optimization seems to work only once. When I closed the file and I reopen it, it become impossible to launch a further optimization.
I took a screen shot that you will find at: https://mycore.core-cloud.net/index.php/s/JGvZhIn8jYbdXMR
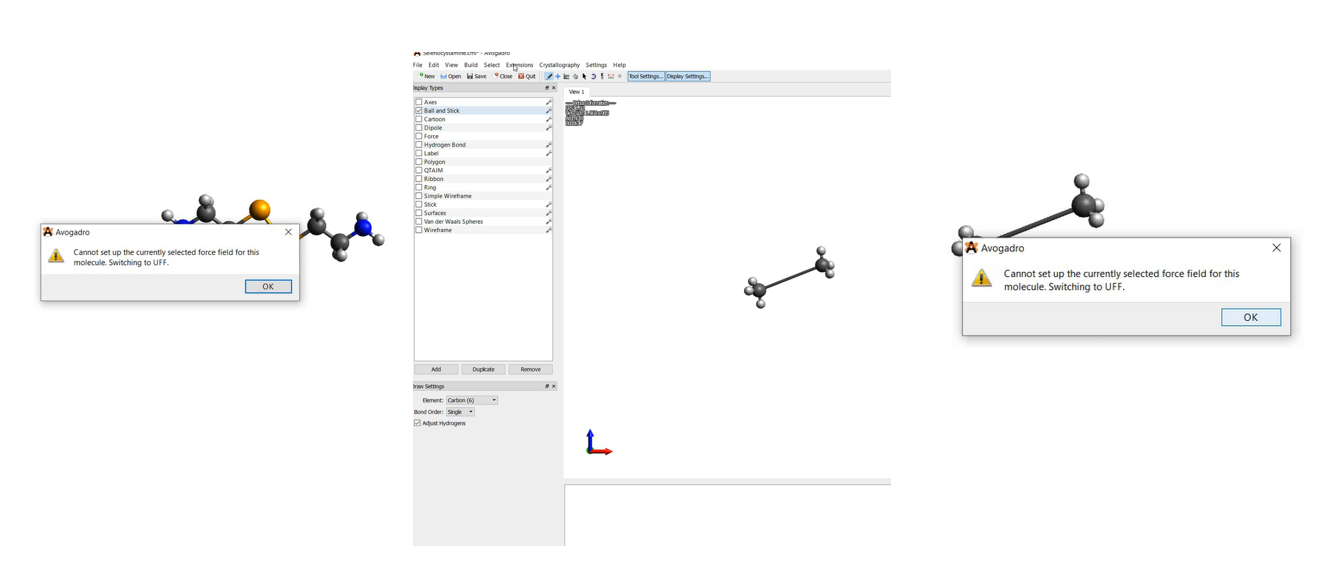
I encountered the same bug as well. I was editing a molecule containing Se. Even after I manually choose “Set up force field” and selected UFF, it says it’ll revert back to UFF and does not do any optimization.
What I did is clicking “quit” and then restart from the scratch. Somehow it just worked.
Then I click “Optimize Geometry”, but the message "Cannot set up the currently selected force field shows up.
To see if it’s something going on with the Se. I erase everything and draw an ethane.
Curiously enough, I still get the same message when I run “Optimize geometry”.
Of course, if I start over with a fresh new file it does not happen. So my guess is that something that is saved in the background got messed up and the bug status is not updated.
There’s definitely a bug. On the other hand, we can’t support Avogadro 1.2 anymore. So I’ll test as I’m working on the new optimization framework in Avo2.
I had the same problem and solved it by first opening Avogadro exe and opening the .cml file from the file tab instead of just clicking the file on my desktop. I’m using windows 11.