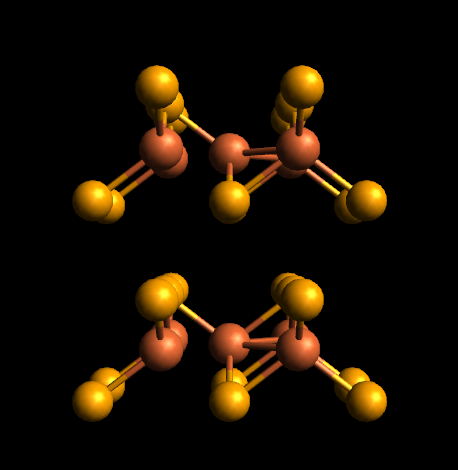
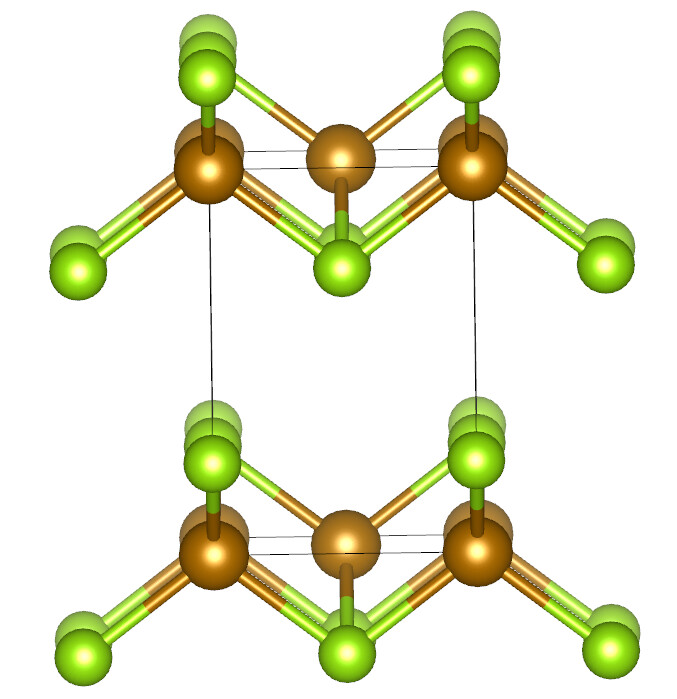
In Avogadro, bonds are perceived based on distance (e.g., are the Fe-Fe distances small relative to the covalent radii). So if the atoms are close enough, they will get a bond.
Vesta likely has a different method (e.g., ignoring metal-metal bonds)
@yujin4169 The syntax of .cif permits the description of bonds between atoms. That is, you may find a block with explicit definition of the the atoms bond together, distance (in Angstroms), and (local) symmetry e.g.,
but such a block is not mandatory though the assignment of a bonding between atoms however can be helpful in the course of establishing (solving) and improving (refining) a crystallographic model. As an illustration, see for instance a contemporary model (COD 4002152, published 2015) vs COD 1010298 (of data published in 1933). In addition to this – contrasting to a .sdf file and its bond block – a typical .cif file does not store information about bond order either. Crystallographers still can recognize if a bond is unusual by length by comparison with compilations like volume C, chapter 9.5 of the (International Tables for Crystallography) Typical interatomic distances: organic compounds (an apparently public link). Modern programs dedicated to the field are aware about these data, too. For more information about the .cif files’ syntax, see e.g., IUCr’s core .cif dictionary.
The .xyz file format neither retains information about bond border, nor about which atoms are close enough to form a bond. They only state the number of atoms and their individual atomic coordinates as mandatory content, plus some (general) comment about the structure file if wanted, or else the line remains empty. In contrast to a .cif, its underlying coordinate system always is Cartesian.
The attachment below lists all public entries of the COD which contain only Fe and Se as elements.