I have an xyz file containing some atoms and their coordinates. Is there an easy way to label the atoms in order of appearance? Even after toggling atom labels in avogadro, exporting to an xyz file drops the atom label numbers. For example, I’d like to convert my xyz file from this:
atom x y z
C # # #
H # # #
H # # #
H # # #
C # # #
H # # #
H # # #
H # # #
And change it to something like this:
atom x y z
C1 # # #
H1 # # #
H2 # # #
H3 # # #
C2 # # #
H4 # # #
H5 # # #
H6 # # #
I’m fairly certain that the xyz format never includes the atom label numbers, so if anyone knows of a simple way to do this after exporting, I’m all ears.
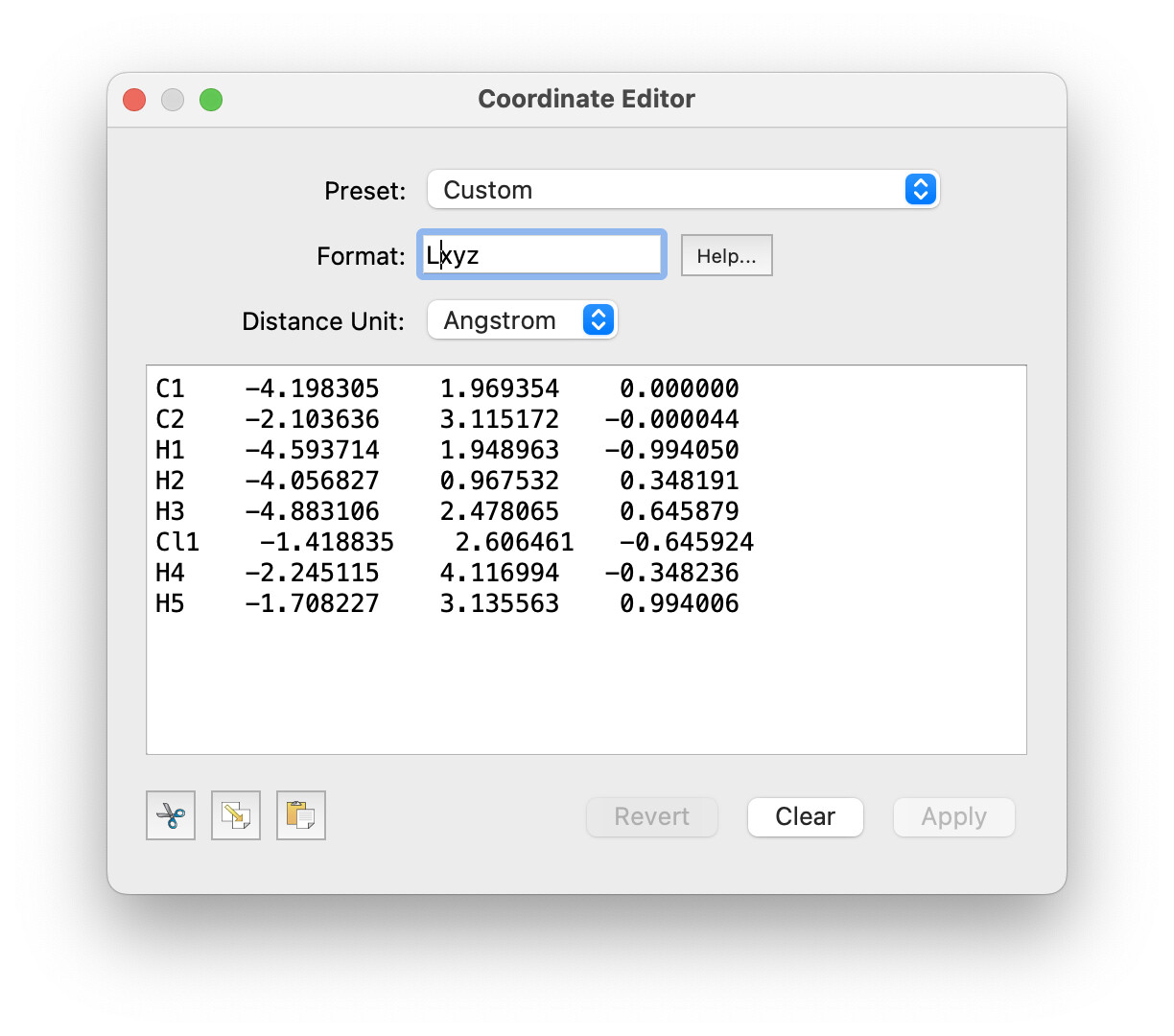
No, the XYZ format doesn’t support this. But you can copy / paste from the coordinate editor. You can set a custom format for the coordinate display, e.g.:
Yep this seems to work fine if you just want a counting total of the number of atoms. Although, in the example you show, I would need the first H to be labeled “H1” since it’s the first hydrogen. I agree, there is probably a simple python script to perform this.
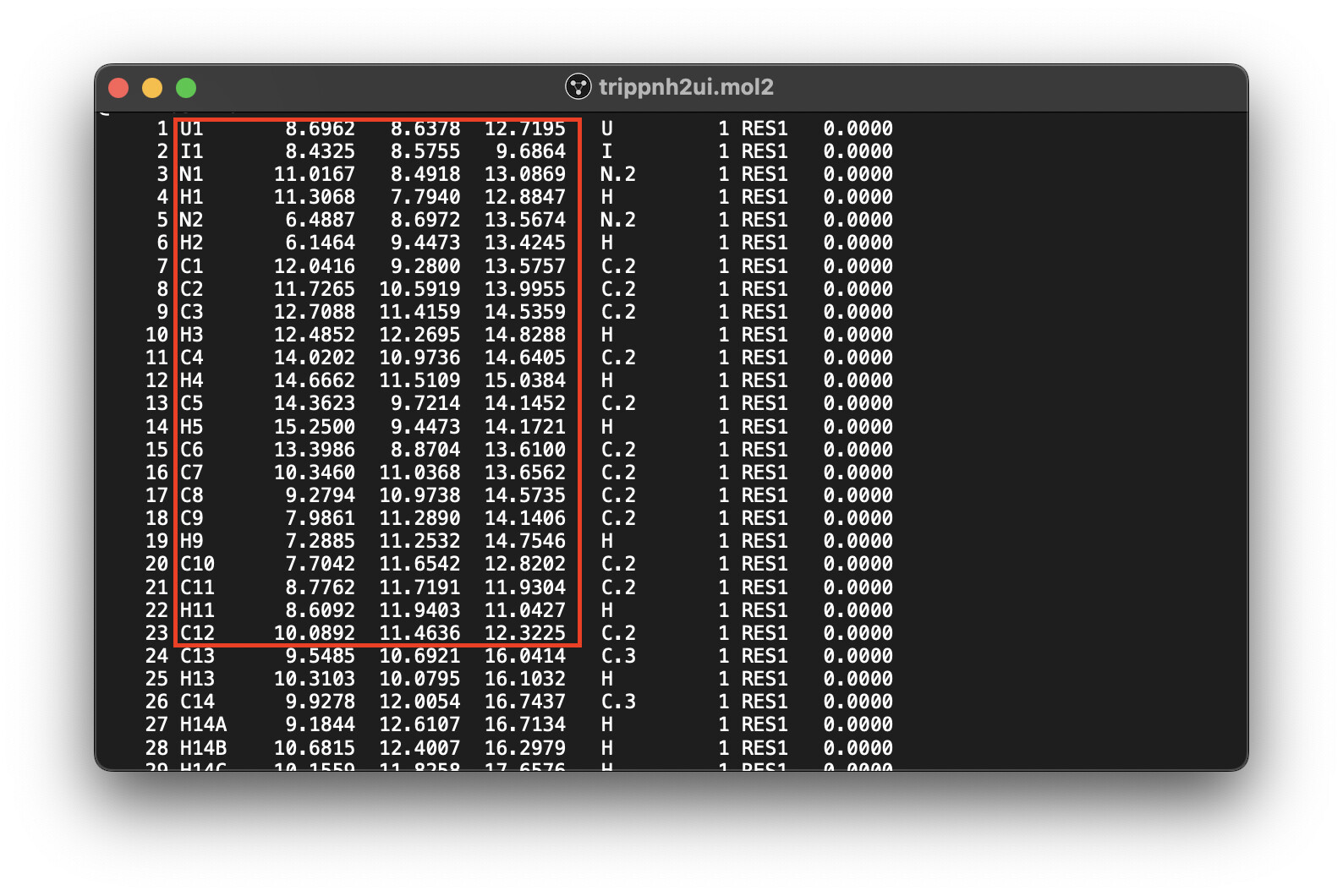
One solution (although a bit messy) is to export the file as a .mol2 (distinctively not .mol). I had to use mercury for this. mol2 includes a section where the atoms are listed and numbered like I needed. You then just have to copy/paste the part you want from the .mol2 text file. See pic below of highlighted area.
No other export format that I could find seems to include the atoms listed and numbered in this way, which seems like a strange oversight considering there are multiple computational programs that need a numbered list of atoms just like this.
Additionally, I might just be silly and missing something simple.
@ianbackwardsnai Column 1 of your example displays a pattern of atom labels which is sometimes seen in crystallography (well, by Mercury as a software, I think about CCDC’s Mercury). In the course of structure solution, the positions of non-hydrogen atoms such as carbon, nitrogen are determined first. Hydrogens might be seen in the electron map (especially for diffraction experiments with neutrons), but often not (more common for X-ray diffraction). Often they are added on idealized (riding) positions, programs like ShelX (small molecule crystallography) «know» the typical geometries and (temperature dependent) distances to complete the model eventually refined. For easier «bookkeeping», hydrogens’ labels the carry a label relating to the non-H atom, e.g. H12 is not the twelfth hydrogen assigned, but because it is bound to e.g. a nitrogen atom which happens to be labeled by N12. By similar token (cf. lines 27–29 in your screen photo), it often is very much helpful if hydrogens of a methyl group carry an indicator in common, e.g. C14very likely is the root of the three hydrogens H14A, H14B, and H14C with an additional letter when modelling positional disorder, for example. (One often applies the same for fluorine atoms of trifluoromethyl groups, by the way.)
There equally are instances where labeling non-H atoms across a centroid as if there were a centre of inversion is helpful, too. To highlight the pairwise relationship C10 (and its parameters) are “echoed” by C20; for C11 there is C21; for C12 there is C22 on the other side; I assume you get the picture.
So you might loose information if you exchange the labels H14A, H14B, H14C for example by H14, H15, H16 (or down to a plain .xyz file) while actually the fifth column is about the assigned atom type. And as you see, there is enough difference for the program used to provide the listing to discern carbon atoms by C.2, or C.3 – think of C(sp^2) vs C(sp^3) with different typically observed bonding geometries, and bond lengths to atoms close by. In absence of an explicit bond block (as in .sdf files see here), programs access the atom type to draw a strut between two atoms to symbolize a bond. (Additional criteria such as empiric rules of valence and van der Waals radii apply.)