I would like to be able to specify an inter-nuclear axis as one of the Cartesian axes. This would help when interpreting a series of calculations where the outputs might be arbitrarily rotated. I could specify the axis, and use the center and align tools. Specifying some bond as a Cartesian axis would be one way to improve the consistency and professionalism of images generated via Avo2.
Yes, I haven’t had a chance to port the align tool, which would allow you to specify an atom as the center, and another atom along one of the Cartesian axes. (In Avo2, this could be made into a script command as well.)
My hope was to finish that off this week - we need it in the group anyway.
Okay, I have a pull request that adds back the align tool, as well as two new script commands:
centerAtom which expects an atom index
alignAtom which expects a second atom index and an axis (e.g. “x”)
I need to work on docs and examples, but it should get merged tomorrow. (Then back to work on the force fields.)
Not sure I am using the message passing right.
I’ve tried:
print("aligning atoms")
sendMessage("centerAtom", {"index": 3})
sendMessage("alignAtom", {"index": 12, "axis": "y"})
and
print(“aligning atoms”)
sendMessage(“centerAtom”, {“id”: 3})
sendMessage(“alignAtom”, {“id”: 12, “axis”: “y”})
to no effect. I’ve inserted them into the MO generation script you shared. I’m sure its just that I am misunderstanding how to use the commands. Could you correct my script?
Nope, it’s nothing you’re doing wrong.
I found a ![]() last night / today … the script commands that I added to tools don’t seem to connect. Script commands added to “extensions” (like the surface generation menu commands) work just great. But the align tool … and code I added to the navigate tool to rotate / zoom the scene… nope.
last night / today … the script commands that I added to tools don’t seem to connect. Script commands added to “extensions” (like the surface generation menu commands) work just great. But the align tool … and code I added to the navigate tool to rotate / zoom the scene… nope.
It’s going to drive me nuts. The tools get the signal, but nothing actually happens.
Since I want to get 1.98 out Really Soon Now™ I’ll hack something together.
Ah, ok. That sounds good, I will keep playing with surface gen. I am going to leave a comment in that thread now to keep like with like.
Okay, it took like a month, but with the latest commits to both avogadroapp and avogadrolibs, the tools now get script commands. (The app was saving the tool before it ever got attached to the scene or the molecule.)
So yes, you now should be able to:
sendMessage("centerAtom", {"id": 3})
sendMessage("alignAtom", {"id": 12, "axis": "y"})
I have been attempting to use the align tool in multiple Avogadro versions. Yet, after selecting the atoms I would like to align, selecting the axis, and clicking align - nothing happens.
This does not seem to be an issue on mac but is on windows.
Any guidance would be appreciated, thank you !
Well, I definitely have students in the lab using it on Windows with Avogadro2 1.102, etc.
It would help a bunch to know what version(s) you’re using. And maybe take a screenshot of the Atomic Coordinate Editor… before and after you click the “Align” button.
For example, the atom #1 (Nitrogen in this case) is now at the origin, and atom #2 (Carbon in this case) is now at 0,0, 3.9331Å
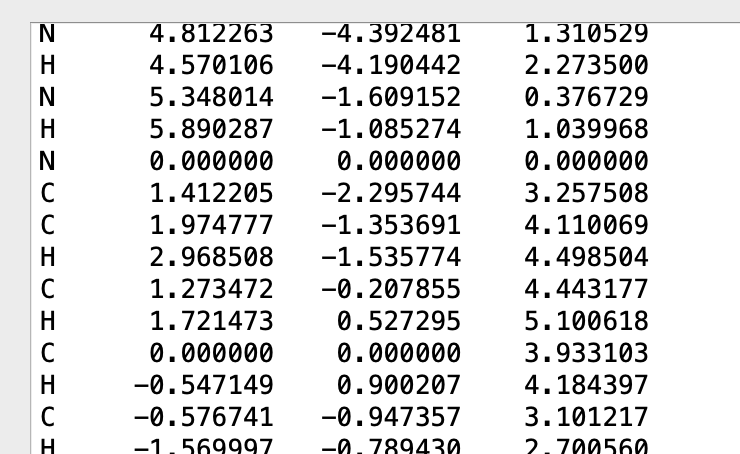
Hi ghutchis, I was about to delete my query as all good, I have managed to get it working with v0.9.3 ! Thanks for the swift reply nonetheless.