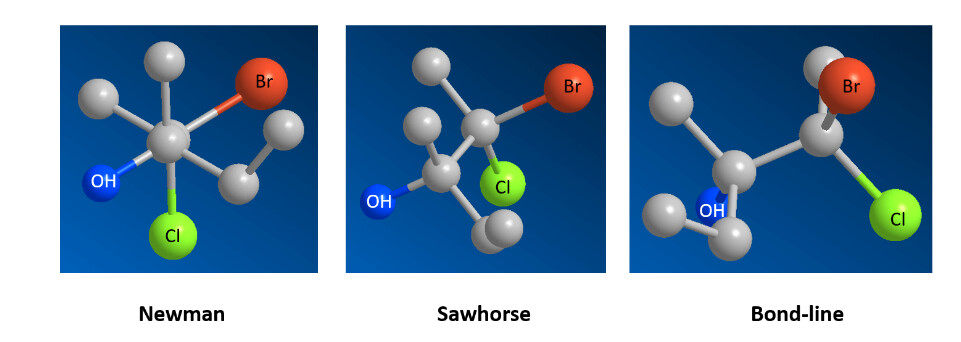
I have a suggestion for a new feature related to this GitHub issue. I’m not sure if it already exists, if not, I think adding bond-line, Fischer, and Newman projections could be a good idea. I’d be happy to help on this feature.
I guess you’re looking for something that will compute appropriate rotations for the modelview? I mean, it’s already pretty trivial to rotate to these projections (or others) as you want.
I suspect the trick would be to compute the “main axis” for the Newman and rotate to find two substituents “up” and “down.”
Some of these seem like they would be hard for anything beyond a simple example molecule though.
For general rotation to see different perspectives I agree that it’s most easily done with the mouse, and as you say, computing these things could be hard.
And a “bond-line” projection doesn’t seem to make any sense to me at all.
The thing is @perminder-17, a projection is just a way of depicting a molecule in 2D to demonstrate stereochemistry, not an actual structure. So what is the meaning of a “projection” in a 3D context? Should applying a projection in Avogadro just adjust the view to give an approximation of the projection, or should the atoms be moved appropriately? A sawhorse projection usually has the bond elongated, for example, so that the substituents don’t clash. And a Newman projection of an eclipsed rotamer is traditionally shown not as the actual eclipsed form but ever so slightly staggered, so the groups at the rear can be seen. But would users really want or expect the atoms to move when applying a projection? Because if they don’t move, you are just rotating the view, which as @ghutchis says, is already trivial to do.
It could maybe make some sense if something like that was added to the pane of an appropriate tool. A Newman projection option could go under either the Align Tool or the Bond-Centred Manipulation Tool, for example, since in both you are selecting two atoms/a bond. Adjustment of the dihedral is already part of the latter, so maybe that’s a good fit. Equally, a Newman projection is essentially doing an alignment along the z-axis like the Align Tool does, just that the z-axis is then set to perpendicular to the screen and the groups should probably be rotated too if necessary to get one of the classic rotamers.
If I was to run with the idea though, being able to select the atoms of a six-membered ring and then get it quickly adjusted to give a classic chair (or boat or twist-boat etc.?) or, if it’s a monosaccharide, a Haworth projection, would be nice too.
Going even further, for sugars it would be ridiculously cool if you could select “Fischer projection” for a cyclic monosaccharide and see the bond break, and the structure rotate until it forms the appropriately flattened chain. And the reverse. Indeed, being able to study a Fischer projection from different angles would be a useful demonstration of why they are unrealistic. Along the same lines, an animated furanose <> pyranose interconversion would be sweet. But the paths for all these would be complex and would have to be guided by force fields.
Certainly a few quick, automatic projections could be cool for educational purposes if they are contingent on relevant atoms being pre-selected. Maybe a “Projection Tool” would be an idea – or maybe it’s the kind of thing that would be better as a Python plugin.
It helps some students to see the orientation of the “front” and “back” of the Newman projection.
Yes, adding a “view along bond” with different options might be a useful tweak to the bond-centric manipulate tool.
But so far as I can tell from Google, isn’t “bond-line” just a way to refer to what we’d consider the normal 2D representation of a molecule?