Which shows that it’s the double back slash:)) I think I have got my menus back now by adding a few of these back slashes. Let me check whether it really works as expected and get back to you.
I still have to run the calculation of the type I started all this activity for - geometry optimisation of multiply protonated peptides, but at least for a neutral molecule all seems to work fine at the moment.
Excellent! Sorry it took so long. Let me know if you have any more issues.
I’ll get a fix out today for the binary path issue. I’d then recommend upgrading the plugin to v0.8.1 from whatever you have (probably 0.6).
I don’t know if you know this or not but to set the charge you’ll need a recent (last week or two) nightly build of Avogadro and then you can change the overall charge in Analysis > Properties > Molecular.
And my top tip would be to basically always use Opt + Freq rather than Optimization because it will squash out any negative frequencies by automatically restarting for you until you get a true minimum.
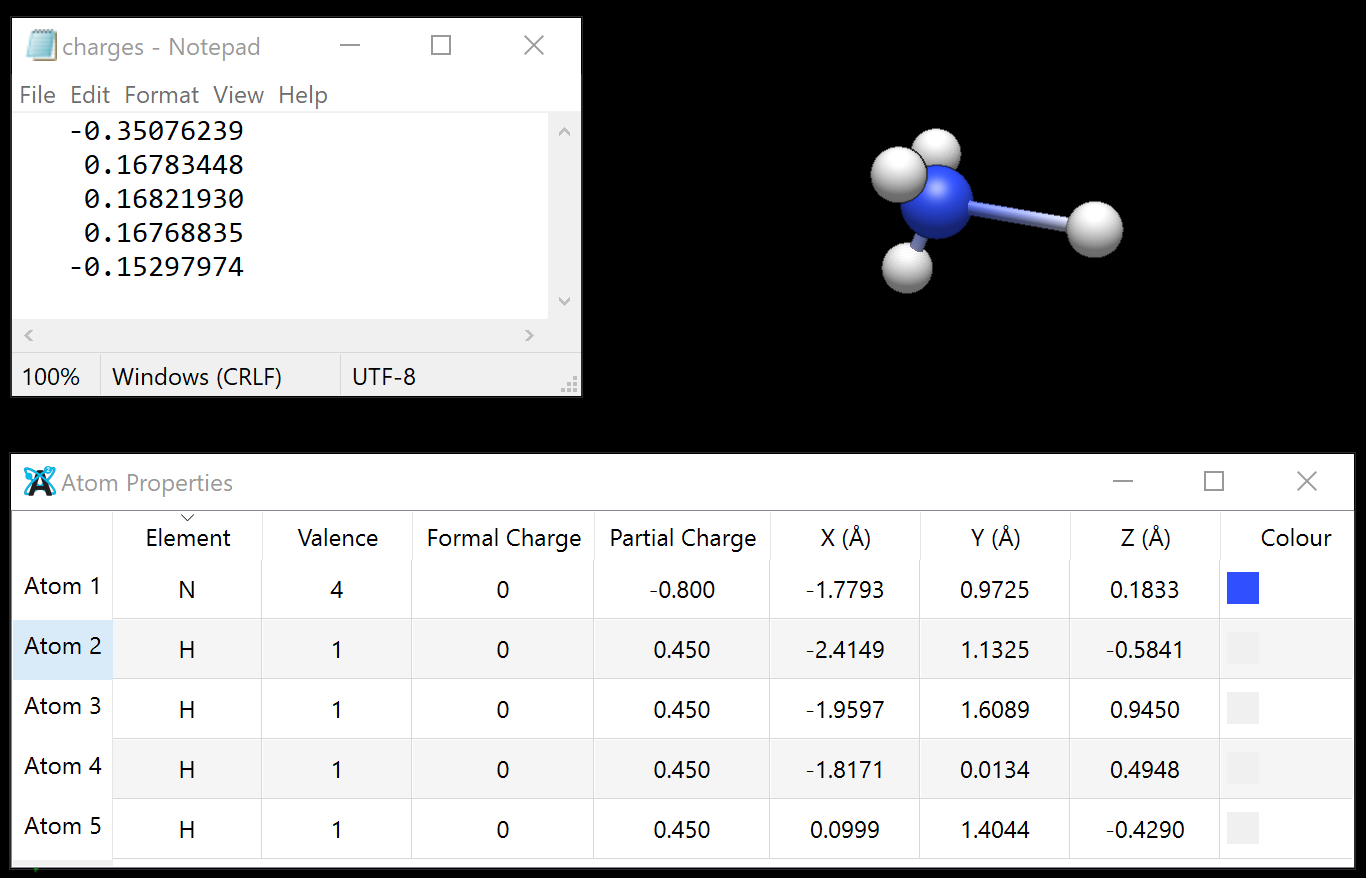
Thanks Matt! Previously, I was advised to set the atomic charge via the atomic properties menu. I tried to see how it works now with the simplest case - ammonia. If I set the charge on the 4-valent N in NH4 to +1, and then run the xtb optimisation, I get an unreasonable geometry and zero net charge - apparently, xtb does not understand that the system is +1 overall despite the manual change in the atomic properties table. That table, however, does change following the xtb optimisation and keeps the overall +1 charge:
It’s a known current bug, it’s useful to know that it affects Windows too.
Grab it from here for now and manually install it (just replace the current C:/Users/You/AppData/Local/OpenChemistry/Avogadro/commands/avo_xtb folder with the newly downloaded one).
Thanks a lot! This does produce the tetrahedral NH_4^+ - success! Although the absolute energy is somewhat strange - different by an order of magnitude from the Hartree-Fock energy. Maybe this is because the core electrons are not considered at all. Also the energy difference between the optimised NH3 and the optimised NH4^+ is smaller by a factor of ~4 than the literature value of the gas phase proton affinity. Maybe a bit naively, I thought this would be a reasonable approximation to the Hartree-Fock solution with frozen core which in its turn should be OK as a first approximation for the gas phase proton affinity.
Yeah, I don’t think you can compare GFN2 total energies to HF energies.
They do have calculations of Fukui indices - and they use --lmo to get localized MOs for their protonation / deprotonation analysis with CREST.
One warning - we found in our pKa ML model QupKake that the GFN2 proton barriers are way too small. It’s actually in the SI notes in the original GFN2 paper, but not otherwise discussed.
So I’d be a bit wary about proton affinity from GFN2 except in a qualitative trend.
Thank you very much, this is very useful to know! The fact that they derive their protonation patterns from the wavefunction rather than from the energy explains a lot. I wonder whether the Fukui indices could be available via the plugin? I understand that at the moment, CREST as a whole is available, but not under windows… Thanks again!
That’s definitely a limitation that’s worth knowing about – any idea why it fails in that specific area?
Yeah, I only really put the Energy command in the plugin because it’s kind of expected. Sensible geometries, frequencies, and ensemble generation, and the calculation of properties that follow from those, are I think where it shines.
You’re not the first to request it! Since xtb calculates them regardless it’d be fairly easy to support them. @ghutchis – is it possible to pass them to Avogadro in the CJSON, and if so with what syntax?
Yes, the plugin also acts as an interface to CREST, but sadly they only make binaries available for Linux. I’m not sure it’s even possible to compile it yourself on Windows.