Hi everyone,
I want to look into improving the general molecule editing workflow within Avogadro 2 and based on my experience so far, the manipulation tool is currently implemented in a rather rudimentary way.
Trying to be a good Open Source citizen I figured I might give a shot at improving the situation myself, but before I dive into the code, I wanted to present my ideas here and gather some feedback on them first.
The first improvement that I have in mind would be to introduce modification “gizmos” as is often encountered in other 3D editors (e.g. game engines). See for instance

Then for actually performing any kind of manipulation (be it rotation or translation) one would have to use these gizmos.
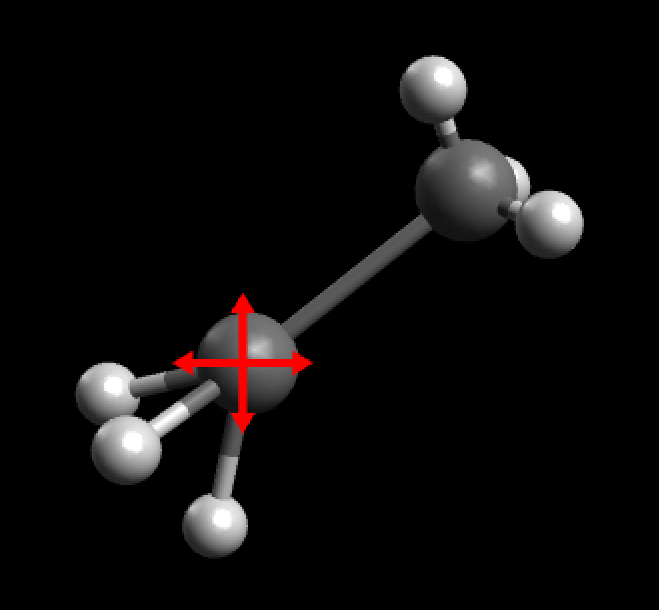
In addition, I think the center of rotation should
a) be displayed
b) be customizable
As a further extension I thought that it could be beneficial to allow the user to define arbitrary help-lines and/or -planes (e.g. by selecting two atoms and then creating a line that connects those two or by selecting three atoms and creating the plane which they form) which can be used to restrain the manipulation (e.g. keep atoms within the plane, move them along the line, rotate around axis defined by this line, etc.). Note however that these helper geometries should also be definable by arbitrary points and not necessarily by existing atoms or bonds (that way one could e.g. define a rotation axis that goes through the center of mass between three atoms, perpendicular to the plane that they span).
Furthermore, I am wondering whether it really makes sense to keep the bond-centric manipulation tool as a separate tool. I could imagine that it might be beneficial to unify all currently available editing functionality into a single tool and then extend that tool by the things mentioned above.
The result would be a single modification tool that should cover free-form modifications as well as bond-centric manipulations.
I believe that for a user this would greatly improve the current workflow.
While we’re at it, I think there would also be value in essentially assimilating the functionalities of the select tool into this unified tool as well.
By using transformation gizmos, we’d have to have a way of selecting atoms and bonds anyway and having to switch the used tool for this every time seems rather cumbersome.
So I think in the end, what I would propose is a modification tool that combines and extends the functionality of the current tools manipulate, select and bond-centric manipulation.
Concretely I would imagine a typical workflow to be the following:
- Click on an atom. This selects the atom and shows the combined translation/rotation gizmos (though for a single atom rotation probably doesn’t make sense, so we could restrict ourselves to showing only translation gizmos)
- Alternatively drag the mouse to select a group of atoms. Show the translation/rotation gizmos and use the center of mass as the default rotation pivot point (and visibly mark it!)
- From here the user could either drag on the translation gizmo (arrow) to move the entire group of atoms along the respective axis or drag on the center of the gizmo to freely move the selection parallel to the camera’s plane
Alternatively the user could drag on one of the rotation gizmos in which case the selection is rotated around the respective respective axis going through the rotation pivot
Furthermore it would be possible to move the rotation pivot itself by either dragging it through space or by letting it snap to a specific atom or bond - There should be the possibility of changing the used axis system for the abovementioned processes. The choices should include “global space” aka the cartesian coordinate axis, “local space” which should somehow be defined by the bonds coming out of the selected atom (though there is some ambiguity if there are more than three bonds) as well as some way of essentially freely defining the coordinate system (similarly to how the manipulation plane can currently be changed in the bond-centric manipulation tool).
- If instead of an atom, the user clicks on a bond, select that bond instead and provide options for changing the bond-length, the bond- and/or dihedral angle (aka: Essentially reuse what the bond-centric manipulation tool currently does)
The ability to create helper lines and planes might also be worth to keep around as a separate tool as these things could also be interesting for e.g. the measure tool (measure the angle between two arbitrary lines that the user created, without those necessarily having to be defined as passing through two atoms).
Please let me know what you think of this idea and/or if this needs more clarification.