HI-
I’m running
Avogadro version: 1.96 on MacOS 12.4 (Monterey) and trying to setup Avogadro to do optimization using GAMESS.
What I had hoped to do was setup a molecule, then use Avogadro to create a job file for GAMESS that (presumably) would be dispatched by Molecule. My best attempts at configuring Molecule to execute GAMESS simulations is not working. With Molecule running, submitting a GAMESS job through Avogadro via Molecule ends with the message: “Please select the target program from the Queue and Program list” even though there is nothing to select in the Q-and-P options on the “Submit GAMESS Calculation” dialogue box.
I’ve included screen-caps (below) of what I am seeing, but does anyone have any pointers on how to properly set this up?
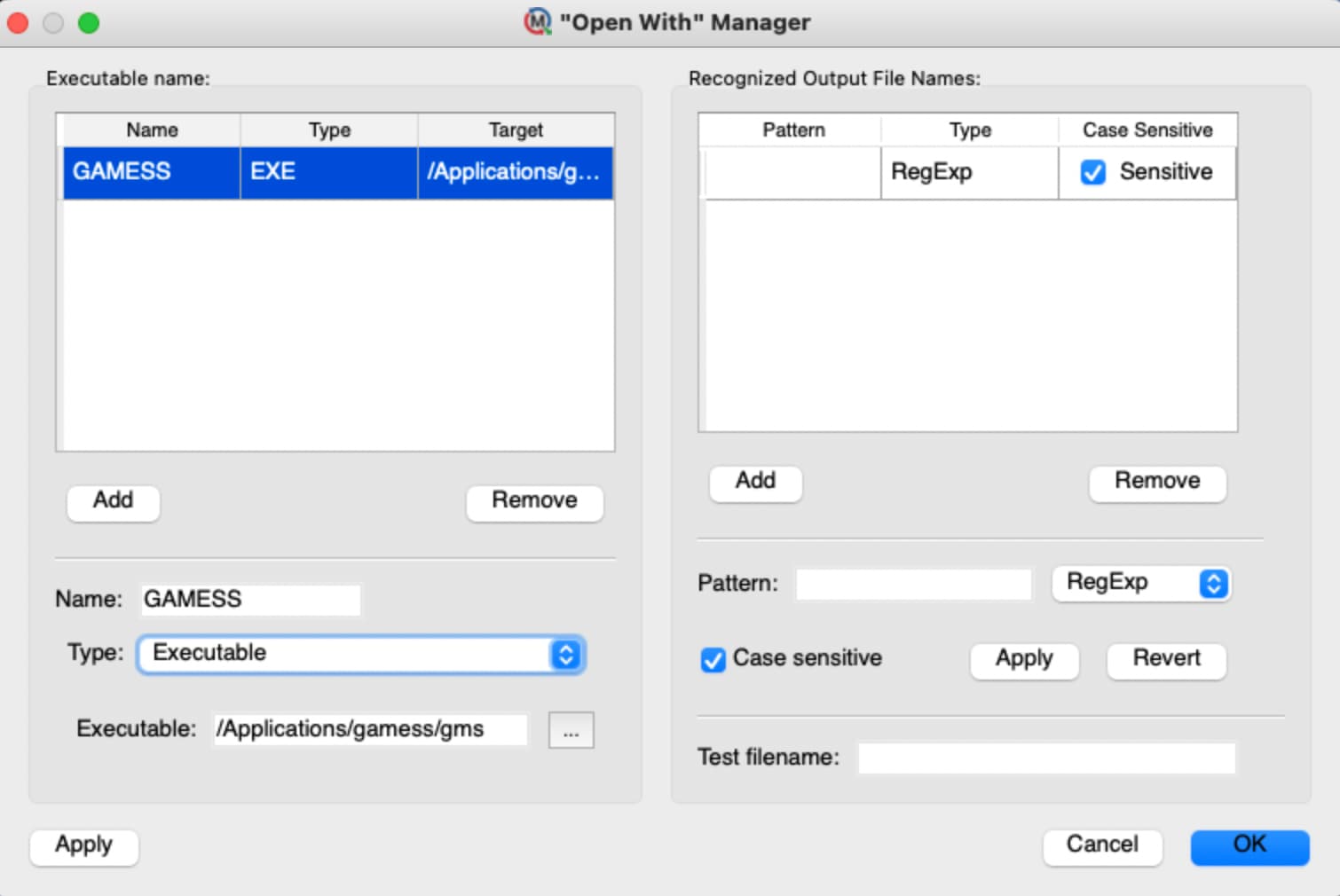
My Molecule Setup
Best wishes and thanks,
Kevin
MoleQueue needs some real love and/or help - in the way of documentation and tutorials. At the moment, it’s sort of a second thought because I’m trying to finish bits of Avo2. There’s no one specifically working on MoleQueue.
I think my first question is where you’ve installed the gms script on your Mac?
Thanks very much for chiming in here. Am a fan of Avo2 and look forward to seeing more of your work. 
Yes, I’ve installed gms in the default location on my Mac; but have the sense that Molequeue is asking me to describe that location to it (and or to Avogadro) and somehow I am not making myself clear. I did my best with the interfaces provided and plugged in an absolute path the executable in what I thought was the correct dialogue box on Molequeue and have verified that Avogadro can connect to Molequeue; but am still stuck.
FYI – I’ve even successfully run gms optimizations from the command line (outside of Avogadro/Molequeue), but have not gotten the suite to work right.
Maybe I should adapt the approach here while Molequeue is under development and just run scripts to drive gms?
Still unclear to me what kind of gms output files are compatible with Avogadro import. My overall goal here is to visualize orbitals in Avogadro that are computed via GAMESS. Any advice you have would be appreciated.
Kevin
I’ll be up-front that I haven’t used GAMESS in eons. The code looks like it’s reading ATOMIC BASIS SET sections for MOs? I don’t know if that requires a particular keyword in the GAMESS input. (As I said, avogadro-cclib should also read output and supply MO / basis set information if it’s in the output file.)
We can probably get MoleQueue working for you, but in the meantime scripts definitely work.
When you read in output files, and go to Analysis => Create Surfaces… do you see orbitals?
…sorry for the long delay in my reply…
I’ve been able to successfully run optimization jobs through GAMESS on the command line using Avogadro-generated .inp files
Still have not figured out how to get Avogadro to read in the output from GAMESS’s *.dat output file format. Ideally, I’d like to be able to take some of that into Avogadro and see with surfaces via Analysis => Create Surfaces… but Avogadro apparently needs me to somehow tell it what the GAMESS output was.
Any clues you have on this would be welcome.
Thank you!
Kevin
Avogadro has never read .dat files. Both Avogadro and Open Babel (and I believe cclib as well) have always read the .out or .log output files.
1 Like