I am using Avogadro 1.2.0 and I saw (while opening a new file) that
Avogadro can read NWchem output files. I have a frequency calculation
output file (my log file, a text file) from NWChem 6.6 that makes
Avogadro crash when I try to open it.
NWChem also prints 20 xyz files for each mode and a single file that
includes all 20 xyz geometries. I have been able to use Avogadro to
read that single file with the 20 xyz geometries to make a movie of
the vibrational mode but I have not found a way to make a figure that
displays the molecule and arrows to display the vibrational mode.
My questions:
1) Can I use Avogadro to read NWChem output files? Perhaps I am asking
Avogadro to read the wrong output file?
2) Is there a way to use Avogadro to make a figure to illustrate the
vibrational modes?
Avogadro reads all files through Open Babel which supports a wide range of formats, but it’s entirely possible that the NWChem support in OB has not kept up with recent versions. (Many comp. chem programs make subtle but problematic changes in their output files.)
The vibrational mode arrows are read from the output, so it would be a bit tricky to work that backwards from the XYZ files.
I know that Open Babel does read Molden vibrations - if it’s possible to save a Molden file from NWChem, it should have the vibrational results.
@Miroslav_Ilias Would it be possible how NWChem records the results underwent a reorganization?
Though I’m not a user of NWChem, after downloading the file you shared as ...out (i.e. without the additional _SAVED), I attempted to read it with an Avogadro AppImage for Linux from here. Avogadro recognizes it is about the results by NWChem, however reports «Could not find any atomic coordinates. Are you sure this is an NWChem output file?» and stops here.
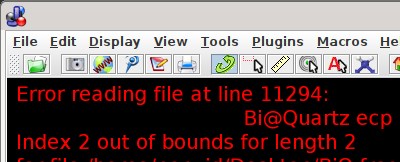
I then turned to Jmol (version 16.1.11 2023-04-19 20:17). This program reports line 11294 as problematic
NWChem is not in Jmol’s list of programs here, and one potential pitfall to test/compare with the test files of the project (e.g. Jmol’s file H2O_1.nwo from here) is their age (NWChem 4.6 vs version 7.2.0 you shared).
It would help to have a bunch of NWChem output files. Since my group doesn’t use it, I don’t have many examples to test.
Reading the geometry is a relatively easy lift - this file had lowercase element symbols:
Importing other parts (e.g., basis, NMR, etc.) will take more time and examples - it’s pretty clear that the NWChem support (which was coded partly with help from the NWChem team) is pretty minimal right now.
Based on these two observations, I’ll check with a current version of Jmol and share an update regarding all 52 files later the day.
If you use NWChem, do you know if there was a substantial change in syntax during the last few years to account for? This is because the repository of Jmol’s reference files on sourceforge appears to me a bit old. On the other hand I remember different file extensions for different releases of Gaussian sometimes requiring an edit before being read e.g., by Molekel, where the project actually provided a converter script between Gaussian 09 to 03.
This should now be in the continuous builds. The following should work:
reading geometry (including conversion of coordinates from Bohr if needed)
reading unit cells / lattice vectors
vibrations
I’ll see what I can do as far as molecular orbitals, reading in multiple geometries, etc. One question is how to handle jobs with multiple steps, different methods, etc. since the energy scales will change dramatically.
@Miroslav_Ilias With the current Jmol release (16.3.37 2025-11-16 11:11), it is possible to read 26 structures out of 52 .out files. See the attached archive. For the other, Jmol struggled with the file syntax and eventually failed to process them. If the screen photos enclosed miss to depict AOs/MOs the computation was about, than this is my fault (because quantum chemistry isn’t my forte).
May your files be used as a reference to update / consolidate the compilation of reference files in the Jmol project?